Powered by Biogen Poland Sp. z o.o.
Powered by Biogen Poland Sp. z o.o.
Tecfidera 120 mg kapsułki dojelitowe, twarde.
Tecfidera 240 mg kapsułki dojelitowe, twarde.
Tecfidera 120 mg kapsułki dojelitowe, twarde
Każda kapsułka dojelitowa twarda zawiera 120 mg fumaranu dimetylu (dimethylis fumaras).
Tecfidera 240 mg kapsułki dojelitowe, twarde
Każda kapsułka dojelitowa twarda zawiera 240 mg fumaranu dimetylu (dimethylis fumaras).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Kapsułka dojelitowa, twarda
Tecfidera 120 mg kapsułki dojelitowe, twarde
Zielono-białe kapsułki dojelitowe twarde, rozmiar 0, z nadrukiem „BG-12 120 mg” zawierające mikrotabletki.
Tecfidera 240 mg kapsułki dojelitowe, twarde
Zielone kapsułki dojelitowe twarde, rozmiar 0, z nadrukiem „BG-12 240 mg” zawierające mikrotabletki.
Produkt leczniczy Tecfidera jest wskazany do stosowania u pacjentów dorosłych oraz u dzieci i młodzieży w wieku 13 lat i starszych z rzutowo-remisyjną postacią stwardnienia rozsianego (ang. relapsing-remitting multiple sclerosis, RRMS ).
Leczenie powinno być rozpoczęte przez lekarza mającego doświadczenie w leczeniu stwardnienia rozsianego.
Dawkowanie
Dawka początkowa wynosi 120 mg dwa razy na dobę. Po 7 dniach dawkę należy zwiększyć do zalecanej dawki podtrzymującej, czyli 240 mg dwa razy na dobę (patrz punkt 4.4).
Jeżeli pacjent pominie dawkę, nie powinien przyjmować podwójnej dawki. Pacjent może przyjąć pominiętą dawkę, tylko jeśli zostanie zachowany odstęp 4 godzin pomiędzy dawkami. W przeciwnym razie pacjent powinien poczekać do planowanego czasu przyjęcia kolejnej dawki.
Tymczasowe zmniejszenie dawki do 120 mg dwa razy dziennie może ograniczyć występowanie działań niepożądanych, takich jak nagłe zaczerwienienie skóry oraz reakcje ze strony układu pokarmowego. Po upływie miesiąca należy wznowić stosowanie zalecanej dawki podtrzymującej, czyli 240 mg dwa razy dziennie.
Produkt leczniczy Tecfidera należy przyjmować z posiłkiem (patrz punkt 5.2). U pacjentów, u których występują działania niepożądane ze strony układu pokarmowego lub nagłe zaczerwienienie skóry, przyjmowanie produktu leczniczego Tecfidera z posiłkiem może poprawić tolerancję leku (patrz punkt 4.4, 4.5 i 4.8)
Szczególne grupy pacjentów
Osoby w podeszłym wieku
W badaniach klinicznych produkt leczniczy Tecfidera stosowano u zbyt ograniczonej liczby pacjentów w wieku 55 lat i starszych, a także u niewystarczającej liczby pacjentów w wieku 65 lat i starszych, aby ustalić, czy reagują oni na produkt inaczej niż młodsi dorośli (patrz punkt 5.2). Biorąc pod uwagę mechanizm działania substancji czynnej, teoretycznie nie ma powodów, dla których konieczne byłoby dostosowanie dawki u pacjentów w podeszłym wieku.
Zaburzenia czynności nerek i wątroby
Produktu leczniczego Tecfidera nie badano u pacjentów z zaburzeniami nerek lub wątroby. Kliniczne badania farmakologiczne nie wskazują na konieczność dostosowania dawki (patrz punkt 5.2). Produkt należy jednak stosować ostrożnie u pacjentów z ciężkimi zaburzeniami nerek lub wątroby (patrz punkt 4.4).
Dzieci i młodzież
Dawkowanie u dzieci i młodzieży w wieku 13 lat i starszych jest takie samo, jak u dorosłych. Aktualnie dostępne dane przedstawiono w punktach 4.8 i 5.1, ale nie można ustalić zaleceń dotyczących dawkowania. Dane dotyczące stosowania u dzieci w wieku od 10 do 12 lat są ograniczone. Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego Tecfidera u dzieci w wieku poniżej 10 lat. Dane nie są dostępne.
Sposób podawania
Podanie doustne.
Kapsułkę należy połykać w całości. Kapsułki ani jej zawartości nie należy kruszyć, dzielić, rozpuszczać, ssać ani rozgryzać, ponieważ powłoczka dojelitowa mikrotabletek zapobiega wystąpieniu podrażnienia przowodu pokarmowego.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Podejrzenie lub rozpoznanie postępującej wieloogniskowej leukoencefalopatii (PML).
Badania krwi/analizy laboratoryjne
Czynność nerek
W badaniach klinicznych u pacjentów leczonych fumaranem dimetylu obserwowano zmiany w wynikach badań laboratoryjnych czynności nerek (patrz punkt 4.8). Kliniczne znaczenie takich zmian nie jest znane. Zaleca się przeprowadzać ocenę czynności nerek (np. oznaczanie kreatyniny i azotu mocznikowego we krwi oraz ogólne badanie moczu) przed rozpoczęciem leczenia, po 3 i 6 miesiącach leczenia, następnie co 6 do 12 miesięcy oraz zgodnie ze wskazaniami klinicznymi.
Czynność wątroby
W wyniku leczenia fumaranem dimetylu może dojść do polekowego uszkodzenia wątroby, w tym zwiększenia stężenia enzymów wątrobowych (≥3-krotnie przekroczona górna granica normy - GGN) i bilirubiny całkowitej (≥2-krotnie przekroczona GGN). Działania niepożądane mogą wystąpić w ciągu kilku dni, po kilku tygodniach lub po dłuższym okresie od rozpoczęcia stosowania. Po przerwaniu leczenia obserwowano ustąpienie działań niepożądanych. Zaleca się przeprowadzenie badania aktywności aminotransferaz (np. aminotransferazy alaninowej [AlAT], aminotransferazy asparaginianowej [AspAT]) i stężenia bilirubiny całkowitej w surowicy przed rozpoczęciem i w trakcie leczenia, zgodnie ze wskazaniami klinicznymi.
Limfocyty
U pacjentów leczonych produktem Tecfidera może wystąpić limfopenia (patrz punkt 4.8). Bezpośrednio przed rozpoczęciem leczenia produktem leczniczym Tecfidera musi być wykonane badanie pełnej morfologii krwi, łącznie z oznaczeniem liczby limfocytów. Jeżeli liczba limfocytów okaże się być poniżej prawidłowego zakresu, należy przed wdrożeniem leczenia dokładnie zbadać możliwe tego przyczyny. Fumaranu dimetylu nie badano u pacjentów, u których już wcześniej występowała mała liczba limfocytów, a zatem u takich pacjentów lek należy stosować ostrożnie. Leczenia nie należy rozpoczynać u pacjentów z ciężką limfopenią (liczbą limfocytów <0,5 × 109/l).
Po rozpoczęciu leczenia co 3 miesiące trzeba wykonywać pełną morfologię krwi łącznie z oznaczeniem liczby limfocytów. Ze względu na zwiększone ryzyko wystąpienia PML u pacjentów z limfopenią zaleca się wzmożony nadzór i postępowanie, jak następuje:
Liczbę limfocytów należy monitorować do czasu ich powrotu do prawidłowych wartości (patrz punkt 5.1). Gdy to nastąpi i gdy brak innych metod leczenia, decyzja dotycząca ponownego wdrożenia leczenia produktem Tecfidera po jego przerwaniu powinna opierać się na ocenie klinicznej.
Badanie obrazowe metodą rezonansu magnetycznego (MRI)
Przed rozpoczęciem leczenia produktem Tecfidera powinien być dostępny wyjściowy wynik badania MRI (zazwyczaj z badania wykonanego w ciągu poprzednich 3 miesięcy) do celów porównawczych. Należy rozważyć potrzebę wykonania kolejnych badań MRI, zgodnie z zaleceniami obowiązującymi w danym kraju lub rejonie. Badanie takie może być przeprowadzane w ramach ściślejszego monitorowania pacjentów, u których stwierdzono zwiększone ryzyko wystąpienia PML. W przypadku klinicznego podejrzenia PML, należy niezwłocznie wykonać badanie MRI w celach diagnostycznych.
Postępująca wieloogniskowa leukoencefalopatia (PML)
U pacjentów leczonych produktem Tecfidera zgłaszano przypadki PML (patrz punkt 4.8). PML jest oportunistycznym zakażeniem wywołanym wirusem Johna-Cunninghama (JCV), które może prowadzić do zgonu lub ciężkiej niepełnosprawności. Stosowanie fumaranu dimetylu i innych produktów leczniczych zawierających fumarany wiązało się z przypadkami PML w przebiegu limfopenii (liczba limfocytów poniżej DGN). Długotrwała umiarkowana lub ciężka limfopenia zwiększa ryzyko wystąpienia PML w związku ze stosowaniem produktu leczniczego Tecfidera. Ryzyka tego nie można jednak wykluczyć u pacjentów z łagodną limfopenią.
Dodatkowe czynniki, które mogą przyczynić się do zwiększonego ryzyka PML w przebiegu limfopenii, są następujące:
- czas trwania terapii produktem leczniczym Tecfidera; przypadki PML wystąpiły po upływie od około 1 roku do 5 lat leczenia, choć dokładna zależność pomiędzy wystąpieniem PML a czasem trwania leczenia nie jest znana,
- istotne zmniejszenie liczby limfocytów T CD4+, w szczególności limfocytów T CD8+, które są ważnym elementem obrony immunologicznej (patrz punkt 4.8) oraz
- wcześniejsze leczenie immunosupresyjne lub immunomodulacyjne (patrz poniżej).
Lekarze powinni ocenić stan pacjentów w celu określenia czy objawy podmiotowe wskazują na zaburzenia neurologiczne. Jeśli tak, należy ustalić, czy są one typowe dla SM, czy też mogą wskazywać na PML.
W momencie wystąpienia pierwszych objawów przedmiotowych lub podmiotowych wskazujących na PML należy wstrzymać stosowanie produktu Tecfidera oraz przeprowadzić odpowiednie badania diagnostyczne, w tym oznaczenie DNA wirusa JCV w płynie mózgowo-rdzeniowym (PMR) metodą ilościowej reakcji łańcuchowej polimerazy (PCR). Objawy PML mogą przypominać nawrotowy rzut stwardnienia rozsianego. Typowe objawy PML są różnorodne, rozwijają się w ciągu dni lub tygodni i obejmują: postępujące osłabienie po jednej stronie ciała lub niezborność ruchową kończyn, zaburzenia widzenia, zmiany w toku myślenia, pamięci oraz orientacji, prowadzące do splątania i zmian osobowości. Lekarze powinni zwracać szczególną uwagę na objawy podmiotowe wskazujące na PML, których pacjent może nie zauważyć. Należy też poradzić pacjentom, aby poinformowali partnera lub opiekunów o stosowanym leczeniu, ponieważ mogą oni zauważyć objawy podmiotowe, których pacjent nie jest świadomy.
PML może występować tylko przy jednoczesnym zakażeniu wirusem Johna-Cunninghama (JCV). Należy wziąć pod uwagę, że nie zbadano wpływu limfopenii na dokładność testu na oznaczenie miana przeciwciał anty-JCV w surowicy u pacjentów leczonych fumaranem dimetylu. Należy również przypomnieć, że ujemny wynik tego testu (w przypadku prawidłowej liczby limfocytów) nie wyklucza możliwości późniejszego zakażenia wirusem JCV.
Jeśli u pacjenta wystąpi PML, należy całkowicie odstąpić od stosowania produktu leczniczego Tecfidera.
Wcześniejsze leczenie immunosupresyjne lub immunomodulacyjne
Nie przeprowadzono badań oceniających skuteczność i bezpieczeństwo produktu Tecfidera w przypadku zmiany z leczenia innymi lekami modyfikującymi na leczenie produktem Tecfidera. Wcześniejsze leczenie immunosupresyjne może przyczynić się do wystąpienia PML u pacjentów leczonych fumaranem dimetylu.
Przypadki PML zgłaszano u pacjentów leczonych wcześniej natalizumabem, przy czym PML stanowi znane ryzyko związane z jego stosowaniem. Lekarze powinni mieć świadomość, że przypadki PML występujące po niedawnym zaprzestaniu stosowania natalizumabu mogą nie wiązać się z limfopenią.
Ponadto większość potwierdzonych przypadków PML podczas stosowania produktu leczniczego Tecfidera wystąpiła u pacjentów poddawanych wcześniej leczeniu immunomodulacyjnemu.
Zmieniając inny lek modyfikujący przebieg choroby na produkt Tecfidera, należy uwzględnić okres półtrwania i mechanizm działania odstawianego leku, aby uniknąć addytywnego wpływu na układ odpornościowy i jednocześnie zmniejszyć ryzyko nawrotu stwardnienia rozsianego. Zaleca się wykonanie pełnej morfologii krwi przed wdrożeniem leczenia produktem Tecfidera oraz regularnie przeprowadzać to badanie w trakcie leczenia (patrz powyżej: Badania laboratoryjne/badania krwi).
Ciężkie zaburzenia nerek lub wątroby
Produktu leczniczego Tecfidera nie badano u pacjentów z ciężkimi zaburzeniami nerek lub wątroby, a zatem produkt należy stosować ostrożnie w tej grupie pacjentów (patrz punkt 4.2).
Ciężka czynna choroba układu pokarmowego
Produktu leczniczego Tecfidera nie badano u pacjentów z ciężką czynną chorobą układu pokarmowego, a zatem produkt należy stosować ostrożnie w tej grupie pacjentów.
Nagłe zaczerwienienie skóry
Nagłe zaczerwienie skóry stwierdzano u 34% uczestników badań klinicznych leczonych produktem Tecfidera. U większości pacjentów, u których wystąpiło, objaw ten miał nasilenie łagodne lub umiarkowane. Z danych pochodzących z badań z udziałem zdrowych ochotników wynika, że mediatorem nagłego zaczerwienienia skóry związanego ze stosowaniem fumaranu dimetylu jest prawdopodobnie prostaglandyna. W przypadku pacjentów z nieznośnym zaczerwienieniem korzystne może być krótkotrwałe leczenie 75 mg kwasu acetylosalicylowego bez powłoczki dojelitowej (patrz punkt 4.5). W dwóch badaniach z udziałem zdrowych ochotników częstość i nasilenie nagłego zaczerwienienia skóry zmniejszyły się w okresie podawania kwasu acetylosalicylowego
U 3 pacjentów spośród 2560 uczestników badań klinicznych leczonych fumaranem dimetylu wystąpiły silne objawy zaczerwienienia skóry, które były prawdopodobnie spowodowane reakcją nadwrażliwości lub rzekomoanafilaktyczną. Te działania niepożądane nie zagrażały życiu, ale doprowadziły do hospitalizowania pacjenta. Lekarzy i pacjentów należy ostrzec o takim ryzyku w przypadku wystąpienia ciężkiej reakcji zaczerwienienia skóry (patrz punkty 4.2, 4.5 i 4.8)
Reakcje anafilaktyczne
Po wprowadzeniu do obrotu zgłaszano przypadki reakcji anafilaktycznej/anafilaktoidalnej po podaniu produktu Tecfidera (patrz punkt 4.8). Objawami mogą być duszność, hipoksja, niedociśnienie tętnicze, obrzęk naczynioworuchowy, wysypka lub pokrzywka. Nie jest znany mechanizm wywoływania reakcji anafilaktycznej przez fumaran dimetylu. Reakcja ta występuje zwykle po podaniu pierwszej dawki, może jednak wystąpić w dowolnym momencie leczenia i może być ciężka oraz stanowić zagrożenie dla życia. Należy poinformować pacjenta, że jeśli wystąpią objawy przedmiotowe lub podmiotowe anafilaksji, należy przerwać stosowanie produktu Tecfidera i natychmiast skontaktować się z lekarzem. Nie należy wznawiać leczenia (patrz punkt 4.8).
Zakażenia
W badaniach fazy III prowadzonych z kontrolą placebo u pacjentów leczonych produktem Tecfidera i u pacjentów otrzymujących placebo częstość występowania zakażeń (60% w porównaniu do 58%) oraz ciężkich zakażeń (2% w porównaniu do 2%) była podobna. Jednakże, ze względu na właściwości immunomodulacyjne produktu Tecfidera (patrz punkt 5.1), jeśli u pacjenta rozwinie się ciężkie zakażenie, należy rozważyć czasowe wstrzymanie leczenia produktem Tecfidera, a przed jego wznowieniem przeprowadzić ponowną ocenę korzyści i ryzyka. Pacjentom przyjmującym produkt Tecfidera należy zalecić, aby zgłaszali lekarzowi wystąpienie objawów zakażenia. Nie należy rozpoczynać leczenia produktem Tecfidera u pacjentów z ciężkimi zakażeniami, dopóki zakażenie nie ustąpi.
Nie obserwowano większej częstości ciężkich zakażeń u pacjentów z liczbą limfocytów <0,8 × 109/l lub <0,5 × 109/l (patrz punkt 4.8). Jeżeli leczenie jest kontynuowane w obecności umiarkowanej do ciężkiej i długotrwałej limfopenii, nie można wykluczyć ryzyka oportunistycznych zakażeń, w tym PML (patrz podpunkt dotyczący PML w punkcie 4.4).
Zakażenia półpaścem
Podczas stosowania produktu Tecfidera zgłaszano przypadki półpaśca (patrz punkt 4.8). Większości z nich nie uznano za ciężkie, ale zgłaszano też ciężkie przypadki, w tym półpasiec rozsiany, półpasiec oczny, półpasiec uszny, półpasiec z powikłaniami neurologicznymi, zapalenie opon mózgowych i mózgu w półpaścu oraz zapalenie opon mózgowych i rdzenia w półpaścu. Te działania niepożądane mogą wystąpić w dowolnym czasie leczenia. Pacjentów przyjmujących produkt Tecfidera należy monitorować pod kątem przedmiotowych i podmiotowych objawów półpaśca, zwłaszcza gdy zgłaszana jest współistniejąca limfocytopenia. W razie wystąpienia półpaśca należy wdrożyć odpowiednie leczenie. U pacjentów z ciężkimi zakażeniami należy rozważyć zaprzestanie leczenia produktem Tecfidera do czasu ustąpienia zakażenia (patrz punkt 4.8)
Rozpoczęcie leczenia
Leczenie należy włączać stopniowo, aby ograniczyć występowanie nagłego zaczerwienienia skóry oraz żołądkowo-jelitowych działań niepożądanych (patrz punkt 4.2).
Zespół Fanconiego
Podczas stosowania produktów leczniczych zawierających fumaran dimetylu w połączeniu z innymi estrami kwasu fumarowego zgłaszano przypadki zespołu Fanconiego. Zespół Fanconiego jest zwykle przemijający, dlatego ważne jest jego wczesne rozpoznanie i przerwanie leczenia fumaranem dimetylu, aby zapobiec wystąpieniu zaburzeń czynności nerek i osteomalacji. Najważniejsze objawy to: białkomocz, cukromocz (przy prawidłowym stężeniu glukozy we krwi), hiperaminoacyduria i fosfaturia (może występować jednocześnie z hipofosfatemią). Progresja może obejmować takie objawy, jak wielomocz, nadmierne pragnienie i osłabienie mięśni proksymalnych. W rzadkich przypadkach może rozwinąć się osteomalacja hipofosfatemiczna z niezlokalizowanym bólem kości, podwyższony poziom fosfatazy zasadowej w surowicy oraz złamania zmiażdżeniowe. Co istotne, zespół Fanconiego może wystąpić bez podwyższonego poziomu kreatyniny ani niskiego współczynnika filtracji kłębuszkowej. W razie wystąpienia niejednoznacznych objawów, należy rozważyć rozwój zespołu Fanconiego i wykonać odpowiednie badania.
Substancje pomocnicze
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) w kapsułce, to znaczy produkt leczniczy uznaje się za „wolny od sodu”
Leki przeciwnowotworowe, immunosupresyjne i kortykosteroidy
Nie badano stosowania produktu Tecfidera w połączeniu z lekami przeciwnowotworowymi ani immunosupresyjnymi, a zatem należy zachować ostrożność podczas ich równoczesnego stosowania. W badaniach klinicznych nad stwardnieniem rozsianym leczenie nawrotowych rzutów choroby krótkimi seriami dożylnych kortykosteroidów nie wiązało się z klinicznie istotnym zwiększeniem częstości infekcji.
Szczepionki
Podczas leczenia produktem Tecfidera można rozważyć jednoczesne stosowanie szczepionek inaktywowanych zgodnie z krajowym kalendarzem szczepień. W badaniu klinicznym z udziałem ogółem 71 pacjentów z RMMS, u pacjentów otrzymujących produkt Tecfidera 240 mg dwa razy dziennie przez co najmniej 6 miesięcy (n=38) lub niepegylowany interferon przez co najmniej 3 miesiące (n=33) wystąpiła porównywalna odpowiedź immunologiczna (definiowana jako >2-krotne zwiększenie miana przeciwciał w stosunku do stanu przed podaniem szczepionki) na toksoid tężcowy i skoniugowaną polisacharydową szczepionkę przeciwko meningokokom grupy C (neoantygen), podczas gdy odpowiedź immunologiczna na różne serotypy nieskoniugowanej 23-walentnej polisacharydowej szczepionki przeciwko pneumokokom (antygen T-zależny) różniła się w obu grupach. Pozytywną odpowiedź immunologiczną (definiowaną jako ≥4-krotne zwiększenie miana przeciwciał) na trzy szczepionki uzyskano u mniejszej liczby pacjentów w obu grupach. Stwierdzono niewielkie liczbowe różnice w odpowiedzi na toksoid tężcowy i polisacharyd pneumokokowy serotypu 3 na korzyść pacjentów przyjmujących niepegylowany interferon.
Brak danych klinicznych dotyczących skuteczności i bezpieczeństwa żywych, atenuowanych szczepionek u pacjentów przyjmujących lek Tecfidera. Stosowanie żywych szczepionek może nieść za sobą zwiększone ryzyko zakażeń klinicznych, a zatem nie należy podawać ich pacjentom leczonym produktem Tecfidera, chyba że zostanie uznane, iż ryzyko wynikające z braku szczepienia przewyższa ryzyko związane ze szczepionką.
Inne pochodne kwasu fumarowego
Podczas leczenia produktem Tecfidera należy unikać jednoczesnego stosowania innych pochodnych kwasu fumarowego (zarówno miejscowo, jak i układowo).
U ludzi fumaran dimetylu jest w znacznym stopniu metabolizowany przez esterazy zanim przedostanie się do krążenia ogólnego, a jego dalszy metabolizm odbywa się za pośrednictwem cyklu kwasów trikarboksylowych, bez udziału układu cytochromu P450 (CYP). Badania in vitro blokowania i indukcji enzymu CYP, badanie p-glikoproteiny ani też badania wiązania fumaranu dimetylu i fumaranu monometylu (głównego metabolitu fumaranu dimetylu) z białkami, nie wykazały ryzyka interakcji.
Wpływ innych substancji na fumaran dimetylu
W badaniach klinicznych potencjalnych interakcji fumaranu dimetylu z interferonem beta-1a podawanym domięśniowo oraz octanem glatirameru, produktów leczniczych powszechnie stosowanych u pacjentów ze stwardnieniem rozsianym, nie stwierdzono, aby zmieniały one profil farmakokinetyczny fumaranu dimetylu.
Z danych pochodzących z badań z udziałem zdrowych ochotników wynika, że mediatorem nagłego zaczerwienienia skóry związanego ze stosowaniem produktu Tecfidera jest prawdopodobnie prostaglandyna. W dwóch badaniach z udziałem zdrowych ochotników podawanie 325 mg (lub równowartości) kwasu acetylosalicylowego bez powłoczki dojelitowej 30 minut przed zastosowaniem produktu leczniczego Tecfidera przez, odpowiednio, 4 dni i 4 tygodnie leczenia nie zmieniało farmakokinetycznego profilu produktu Tecfidera. Należy rozważyć ryzyko związane z leczeniem kwasem acetylosalicylowym przed podaniem go pacjentom z RMMS, stosującym produkt Tecfidera. Nie badano jednak dłuższego (>4 tygodnie) stosowania kwasu acetylosalicylowego (patrz punkty 4.4 i 4.8).
Równoczesne leczenie produktami nefrotoksycznymi (takimi jak aminoglikozydy, leki moczopędne, niesteroidowe leki przeciwzapalne i sole litu) może zwiększać ryzyko działań niepożądanych ze strony nerek (takich jak białkomocz, patrz punkt 4.8) u pacjentów leczonych produktem Tecfidera (patrz punkt 4.4 – Badania krwi/analizy laboratoryjne).
Spożywanie umiarkowanych ilości alkoholu nie zmieniało ekspozycji na fumaran dimetylu i nie wiązało się z nasileniem działań niepożądanych. Należy unikać spożywania dużych ilości wysokoprocentowych napojów alkoholowych (ponad 30% alkoholu objętościowo) w ciągu godziny od przyjęcia produktu Tecfidera, ponieważ alkohol może prowadzić do zwiększenia częstości żołądkowo-jelitowych działań niepożądanych.
Wpływ fumaranu dimetylu na inne substancje
Badania indukcji enzymu CYP in vitro nie wykazały interakcji pomiędzy produktem leczniczym Tecfidera a doustnymi środkami antykoncepcyjnymi. W badaniu in vivo, jednoczesne stosowanie produktu Tecfidera oraz złożonych, doustnych środków antykoncepcyjnych (norgestymat i etynyloestradiol) nie prowadziło do istotnych zmian w ekspozycji na doustne środki antykoncepcyjne.
Nie przeprowadzono badań interakcji z doustnymi środkami antykoncepcyjnymi zawierającymi inne progestageny, jednak nie wydaje się, by produkt Tecfidera wpływał na ich ekspozycję.
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono wyłącznie u dorosłych.
Ciąża
Dostępne są nieliczne dane dotyczące stosowania fumaranu dimetylu u kobiet w okresie ciąży.(od 300 do 1000 przypadków), pochodzące z rejestru ciąż lub spontanicznych zgłoszeń po wprowadzeniu do obrotu. W rejestrze ciąż u kobiet stosujących produkt leczniczy Tecfidera udokumentowano 289 zebranych prospektywnie przypadków kobiet ze stwardnieniem rozsianym narażonych na fumaran dimetylu. Mediana czasu trwania ekspozycji na fumaran dimetylu wynosiła 4,6 tygodnia ciąży, przy ograniczonej ekspozycji także po szóstym tygodniu (44 przypadki). Narażenie na fumaran dimetylu w tak wczesnym okresie ciąży nie wskazuje na większe niż w populacji ogólnej ryzyko wad wrodzonych czy działania toksycznego na zarodek lub płód. Ryzyko dłuższego narażenia na fumaran dimetylu lub narażenia w późniejszych stadiach ciąży nie jest znane.
Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Ze względów ostrożności zaleca się unikać stosowania produktu leczniczegoTecfidera w okresie ciąży. Produkt leczniczy Tecfidera można stosować u kobiet w ciąży, wyłącznie gdy jest to wyraźnie konieczne i gdy spodziewane korzyści dla matki przewyższają ryzyko dla płodu.
Karmienie piersią
Nie wiadomo, czy fumaran dimetylu lub jego metabolity przenikają do mleka ludzkiego. Nie można wykluczyć zagrożenia dla noworodków/dzieci. Należy podjąć decyzję, czy przerwać karmienie piersią, czy przerwać podawanie produktu Tecfidera, biorąc pod uwagę korzyści z karmienia piersią dla dziecka i korzyść z leczenia dla matki.
Płodność
Brak danych dotyczących wpływu fumaranu dimetylu na płodność u ludzi. Dane pochodzące z badań przedklinicznych nie wskazują na to, aby stosowanie fumaranu dimetylu zwiększało ryzyko obniżenia płodności (patrz punkt 5.3).
Tecfidera nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Najczęstszymi działaniami niepożądanymi są nagłe zaczerwienienia skóry (35%) oraz zdarzenia ze strony układu pokarmowego (tj. biegunka (14%), nudności (12%), ból brzucha (10%, bóle w nadbrzuszu (10%)). Objawy te pojawiały się zwykle w początkowej fazie leczenia (głównie w ciągu pierwszego miesiąca). U pacjentów, u których wystąpiło nagłe zaczerwienienie skóry i zaburzenia żołądkowo-jelitowe, objawy te mogą pojawiać się okresowo w trakcie leczenia produktem Tecfidera. Najczęściej zgłaszanymi działaniami niepożądanymi prowadzącymi do przerwania leczenia są nagłe zaczerwienienia skóry (3%) i zaburzenia żołądka i jelit (4%).
W badaniach klinicznych fazy II i III, kontrolowanych placebo, jak i bez grupy kontrolnej, produkt leczniczy Tecfidera otrzymywało łącznie 2513 pacjentów przez maksymalnie 12 lat (łączna ekspozycja równoważna 11318 pacjentolat). Łącznie 1169 pacjentów było leczonych produktem Tecfidera przez co najmniej 5 lat, a 426 pacjentów było leczonych produktem Tecfidera przez co najmniej 10 lat. Obserwacje wynikające z badań bez grupy kontrolnej i z grupą kontrolną są zgodne.
Tabelaryczne zestawienie działań niepożądanych
Działania niepożądane pochodzące z badań klinicznych, badań dotyczących bezpieczeństwa stosowania po wprowadzeniu do obrotu oraz zgłoszeń spontanicznych przedstawiono w tabeli poniżej.
Działania niepożądane przedstawiono zgodnie z terminologią MedDRA oraz klasyfikacją układów i narządów. Częstość występowania działań niepożądanych określono zgodnie z poniższą klasyfikacją:
- Bardzo często (≥1/10)
- Często (≥1/100 do <1/10)
- Niezbyt często (≥1/1000 do <1/100)
- Rzadko (≥1/10 000 do <1/1000)
- Bardzo rzadko (<1/10 000)
- Nieznana (częstość nie może być określona na podstawie dostępnych danych)
|
Klasyfikacja układów i narządów MedDRA |
Działanie niepożądane |
Kategoria częstości |
|
Zakażenia i zarażenia pasożytnicze |
Zapalenie żołądka i jelit |
Często |
|
Postępująca leukoencefalopatia |
Nieznana |
|
|
Półpasiec |
Nieznana |
|
|
Zaburzenia krwi i układu chłonnego |
Limfopenia |
Często |
|
Leukopenia |
Często |
|
|
Trombocytopenia |
Niezbyt często |
|
|
Zaburzenia układu immunologicznego |
Nadwrażliwość |
Niezbyt często |
|
Anafilaksja |
Nieznana |
|
|
Duszność |
Nieznana |
|
|
Hipoksja |
Nieznana |
|
|
Niedociśnienie tętnicze |
Nieznana |
|
|
Obrzęk naczyniowo-ruchowy |
Nieznana |
|
|
Zaburzenia układu nerwowego |
Uczucie pieczenia |
Często |
|
Zaburzenia naczyniowe |
Nagłe zaczerwienienie skóry |
Bardzo często |
|
Uderzenia gorąca |
Często |
|
|
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia |
Nieżyt nosa |
Nieznana |
|
Zaburzenia żołądka i jelit |
Biegunka |
Bardzo często |
|
Nudności |
Bardzo często |
|
|
Bóle w nadbrzuszu |
Bardzo często |
|
|
Ból brzucha |
Bardzo często |
|
|
Wymioty |
Często |
|
|
Niestrawność |
Często |
|
|
Zapalenie żołądka |
Często |
|
|
Zaburzenia żołądka i jelit |
Często |
|
|
Zaburzenia wątroby i dróg żółciowych |
Zwiększona aktywność |
Często |
|
Zwiększona aktywność aminotransferazy alaninowej |
Często |
|
|
Polekowe uszkodzenie wątroby |
Rzadko |
|
|
Zaburzenia skóry i tkanki podskórnej |
Świąd |
Często |
|
Wysypka |
Często |
|
|
Rumień |
Często |
|
|
Łysienie |
Często |
|
|
Zaburzenia nerek i dróg |
Białkomocz |
Często |
|
Zaburzenia ogólne i stany w miejscu podania |
Uczucie gorąca |
Często |
|
Badania diagnostyczne |
Obecność ciał ketonowych w |
Bardzo często |
|
Obecność albuminy w moczu |
Często |
|
|
Zmniejszenie liczby białych krwinek |
Często |
Opis wybranych działań niepożądanych
Nagłe zaczerwienienie skóry
W badaniach kontrolowanych placebo częstość nagłego zaczerwienienia skóry (34% w porównaniu do 4%) oraz uderzeń gorąca (7% w porównaniu do 2%) była większa u pacjentów przyjmujących produkt leczniczy Tecfidera niż placebo. Objaw ten opisywany jest zwykle jako nagłe zaczerwienienie skóry lub uderzenia gorąca, ale może obejmować także inne reakcje (np. uczucie gorąca, zaczerwienienie, świąd i uczucie pieczenia skóry). Zdarzenia te pojawiały się zwykle na początku leczenia (głównie w pierwszym miesiącu). U pacjentów, w których tak się stało, nagłe zaczerwienienie skóry może powtarzać się okresowo w trakcie leczenia produktem Tecfidera. U większości pacjentów z nagłym zaczerwieniem skóry nasilenie takiej reakcji było łagodne lub umiarkowane. Łącznie 3% pacjentów leczonych produktem Tecfidera przerwało leczenie z powodu nagłego zaczerwienienia skóry. Częstość występowania nagłego zaczerwienienia skóry o ciężkim nasileniu, które może przejawiać się w postaci uogólnionego rumienia, wysypki i (lub) świądu, obserwowano u mniej niż 1% pacjentów leczonych produktem Tecfidera (patrz punkt 4.2, 4.4 i 4.5).
Działania niepożądane ze strony żołądka i jelit
Częstość występowania zdarzeń ze strony układu pokarmowego [np. biegunka (14% w porównaniu do 10%), nudności (12% w porównaniu do 9%), bóle w nadbrzuszu (10 w porównaniu do 6%), ból brzucha (9% w porównaniu do 4%), wymioty (8% w porównaniu do 5%) oraz niestrawność (5% w porównaniu do 3%)] była większa u pacjentów przyjmujących produkt leczniczy Tecfidera niż u pacjentów otrzymujących placebo. Działania niepożądane ze strony układu pokarmowego pojawiały się zwykle na początku leczenia (głównie w pierwszym miesiącu). U pacjentów, u których występują zaburzenia ze strony układu pokarmowego, objawy te mogą pojawiać się okresowo w trakcie leczenia produktem Tecfidera. U większości pacjentów z objawami ze strony układu pokarmowego ich nasilenie było łagodne lub umiarkowane. Cztery procent (4%) pacjentów leczonych produktem Tecfidera przerwało leczenie z powodu działań niepożądanych ze strony układu pokarmowego. Ciężkie zaburzenia ze strony układu pokarmowego, łącznie z zapaleniem żołądka i jelit oraz zapaleniem żołądka, obserwowano u 1% pacjentów leczonych produktem Tecfidera (patrz punkt 4.2).
Czynność wątroby
Z danych uzyskanych w badaniach kontrolowanych placebo wynika, że u większości pacjentów z podwyższonymi stężeniami transaminaz wątrobowych wartości te były <3 razy większe niż GGN. Zwiększoną częstość występowania podwyższonego stężenia transaminaz wątrobowych u pacjentów leczonych produktem Tecfidera niż w grupie placebo obserwowano głównie w ciągu pierwszych 6 miesięcy leczenia. Stężenia aminotransferazy alaninowej i asparaginowej zwiększone ≥3 razy górna granica normy obserwowano odpowiednio u 5% i 2% pacjentów otrzymujących placebo oraz u 6% i 2% pacjentów leczonych produktem Tecfidera. W związku ze zwiększonym stężeniem transaminaz leczenie przerwano w <1% przypadków zarówno u pacjentów leczonych produktem Tecfidera, jak i u pacjentów otrzymujących placebo. W badaniach kontrolowanych placebo nie obserwowano zwiększenia aktywności transaminaz ≥3 razy GGN z równoczesnym zwiększeniem stężenia bilirubiny całkowitej >2 razy GGN.
Po wprowadzeniu produktu do obrotu zgłaszano przypadki zwiększenia aktywności enzymów wątrobowych i przypadki polekowego uszkodzenia wątroby (równoczesne zwiększenie stężenia transaminaz ≥3-krotnie przekroczona GGN oraz stężenia bilirubiny całkowitej >2-krotnie przekroczona GGN) w następstwie podania produktu leczniczego Tecfidera, które ustąpiły po przerwaniu leczenia.
Limfopenia
W badaniach kontrolowanych placebo większość pacjentów (>98%) miała prawidłową liczbę limfocytów przed rozpoczęciem leczenia. Po rozpoczęciu leczenia produktem Tecfidera średnia liczba limfocytów zmniejszała się w ciągu pierwszego roku, osiągając następnie plateau. Ogólnie, liczba limfocytów zmniejszała się o około 30% w porównaniu do wartości wyjściowej. Średnia i mediana liczby limfocytów pozostawały w zakresie wartości prawidłowych. Liczbę limfocytów <0,5 × 109 /l stwierdzono u <1% pacjentów przyjmujących placebo i u 6% pacjentów leczonych produktem Tecfidera. Liczbę limfocytów <0,2 × 109 /l wykryto u 1 pacjenta leczonego produktem Tecfidera i u ani jednego pacjenta z grupy przyjmującej placebo.
W badaniach klinicznych (zarówno kontrolowanych, jak i niekontrolowanych) u 41% pacjentów leczonych produktem Tecfidera występowała limfopenia (zdefiniowana w tych badaniach jako liczba limfocytów <0,91 × 109 /l). Łagodną limfopenię (liczby limfocytów od ≥0,8 × 109 /l do <0,91 × 109 /l) zaobserwowano u 28% pacjentów; umiarkowaną limfopenię (liczby limfocytów od ≥0,5 × 109 /l do <0,8 × 109 /l) utrzymującą się przez co najmniej sześć miesięcy zaobserwowano u 11% pacjentów; ciężką limfopenię (liczba limfocytów <0,5 × 109 /l) utrzymującą się przez co najmniej sześć miesięcy zaobserwowano u 2% pacjentów. W grupie z ciężką limfopenią większość stwierdzanych wartości liczbowych limfocytów pozostawała <0,5 × 109 /l podczas kontynuacji leczenia.
Ponadto, po 48 tygodniach terapii produktem leczniczym Tecfidera w ramach niekontrolowanego, prospektywnego badania porejestracyjnego (n=185) u maksymalnie 37% i 6% pacjentów stwierdzono, odpowiednio, umiarkowane (od ≥0,2 × 109 /l do <0,4 × 109 /l) lub istotne (<0,2 × 109 /l) zmniejszenie liczby limfocytów T CD4+, przy czym częściej występował spadek limfocytów T CD8+, do poziomu <0,2 × 109 /l u maksymalnie 59% pacjentów i do poziomu <0,1 × 109 /l u 25% pacjentów.W kontrolowanych i niekontrolowanych badaniach klinicznych pacjentów z liczbą limfocytów poniżej DGN, u których przerwano leczenie produktem Tecfidera, monitorowano pod kątem powrotu liczby limfocytów do wartości DGN (patrz punkt 5.1).
Postępująca wieloogniskowa leukoencefalopatia (PML)
Zgłaszano przypadki zakażenia wirusem Johna-Cunninghama (JCV) powodującego PML w związku ze stosowaniem produktu leczniczego Tecfidera (patrz punkt 4.4). PML może prowadzić do zgonu lub ciężkiej niepełnosprawności. W jednym z badań klinicznych u 1 pacjenta przyjmującego produkt leczniczy Tecfidera wystąpiła zakończona zgonem PML w przebiegu ciężkiej i długotrwałej limfopenii (liczby limfocytów przeważnie 0,5 × 109 /l do < DGN według zakresu referencyjnego określonego przez lokalne laboratorium).
U kilku pacjentów z PML, u których oznaczono liczbę podtypów limfocytów T w momencie rozpoznania PML, stwierdzono zmniejszenie liczby limfocytów T CD8+ do poziomu <0,1 × 109 /l, podczas gdy redukcja liczby limfocytów T CD4+ była zróżnicowana (od <0,05 do 0,5 × 109 /l) i skorelowana bardziej z ogólnym stopniem nasilenia limfopenii (<0,5 × 109 /l do <DGN).
Długotrwała umiarkowana lub ciężka limfopenia zwiększa ryzyko wystąpienia PML w związku ze stosowaniem produktu Tecfidera. Jednak PML występowała też u pacjentów z łagodną limfopenią. Ponadto większość przypadków PML po wprowadzeniu produktu leczniczego do obrotu występowała u pacjentów w wieku >50 lat.
Zakażenie wirusem półpaśca
W związku ze stosowaniem produktu Tecfidera zgłaszano przypadki zakażenia półpaścem. Podczas długoterminowego badania uzupełniającego, w którym leczonych było 1736 pacjentów ze stwardnieniem rozsianym, około 5% z nich zgłosiło jedno lub więcej zdarzeń zachorowania na półpasiec, z których 42% oceniono jako łagodne, 55% jako umiarkowane a 3% jako ciężkie. Czas do wystąpienia objawów od podania pierwszej dawki produktu leczniczego Tecfidera wynosił od około 3 miesięcy do 10 lat. U czterech pacjentów wystąpiły zdarzenia o ciężkim nasileniu i wszystkie te zdarzenia ustąpiły. U większości pacjentów, włączając tych, u których wystąpiło ciężkie zakażenie półpaścem, liczba limfocytów była wyższa niż DGN. U większości chorych ze współistniejącymi liczbami limfocytów poniżej wartości DGN limfopenię sklasyfikowano jako umiarkowaną lub ciężką. Po wprowadzeniu do obrotu większość przypadków zakażenia półpaścem stanowiły przypadki nieuznane za ciężkie, które ustąpiły po zastosowaniu leczenia. Istnieją ograniczone dane na temat bezwzględnej liczby limfocytów (ALC) u pacjentów z zakażeniem półpaścem z okresu po wprowadzeniu do obrotu. Jednak w momencie zgłaszania u większości pacjentów występowała limfopenia umiarkowana (od ≥0,5 × 109 /l do <0,8 × 109 /l) lub ciężka (od <0,5 × 109 /l do 0,2 × 109 /l) (patrz punkt 4.4).
Nieprawidłowe wyniki badań laboratoryjnych
W badaniach kontrolowanych placebo stężenie ciał ketonowych w moczu (1+ lub więcej) było większe u pacjentów leczonych produktem Tecfidera (45%) w porównaniu do placebo (10%). W badaniach klinicznych nie zaobserwowano niepożądanych następstw klinicznych.
Stężenia 1,25-dihydroksywitaminy D zmniejszały się u pacjentów leczonych produktem Tecfidera bardziej niż w grupie placebo (procentowa mediana zmniejszenia stężenia po 2 latach w stosunku do wartości wyjściowej wynosiła odpowiednio 25% do 15%), natomiast stężenia parathormonu (PTH) zwiększały się bardziej u pacjentów leczonych produktem Tecfidera w porównaniu do placebo (procentowa mediana zwiększenia stężenia po 2 latach w stosunku do wartości wyjściowej wynosiła odpowiednio 29% do 15%). Średnie wartości dla obu parametrów utrzymywały się w granicach normy.
W ciągu pierwszych dwóch miesięcy leczenia obserwowano przemijający wzrost średniej liczby granulocytów kwasochłonnych.
Dzieci i młodzież
W 96-tygodniowym, otwartym, randomizowanym badaniu z grupą kontrolną przyjmującą czynny lek, dzieci i młodzież z RRMS (n=7 w wieku od 10 do poniżej 13 lat i n=71 w wieku od 13 do poniżej 18 lat) leczono dawką120 mg dwa razy na dobę przez 7 dni, a następnie 240 mg dwa razy na dobę przez pozostały czas leczenia.Profil bezpieczeństwa u dzieci i młodzieży wydawał się podobny do obserwowanego wcześniej u pacjentów dorosłych.
Schemat badania klinicznego u dzieci różnił się od badań klinicznych kontrolowanych placebo u dorosłych. Z tego względu nie można wykluczyć, że zastosowanie innego schematu badania klinicznego przyczyniło się do wystąpienia różnic liczbowych w zakresie zdarzeń niepożądanych między populacją dzieci i młodzieży a populacją dorosłych. . Te działania niepożądane były zgłaszane u dzieci i młodzieży z następującą częstością (podaną w procentach)
Zaburzenia żołądka i jelit oraz zaburzenia układu oddechowego, klatki piersiowej i śródpiersia oraz działania niepożądane w postaci bólu głowy i bolesnego miesiączkowania zgłaszano częściej (≥10%) w populacji dzieci i młodzieży niż w populacji osób dorosłych:
W niewielkim, 24-tygodniowym, otwartym, niekontrolowanym badaniu z udziałem dzieci i młodzieży w wieku od 13 do 17 lat z RRMS (120 mg dwa razy na dobę przez 7 dni, a następnie 240 mg dwa razy na dobę przez pozostały czas leczenia; n=22), po którym nastąpiło 96-tygodniowe badanie uzupełniające (240 mg dwa razy na dobę; n=20), profil bezpieczeństwa wydawał się podobny do obserwowanego u pacjentów dorosłych.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem: Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C
PL-02 222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu
Zgłaszano przypadki przedawkowania produktu Tecfidera. Objawy opisywane w tych przypadkach były zgodne ze znanym profilem bezpieczeństwa produktu Tecfidera. Nie są znane żadne metody terapeutyczne, które mogłyby przyspieszyć eliminację produktu Tecfidera, jak również nie jest znane antidotum. W razie przedawkowania zaleca się wdrożenie objawowego leczenia wspomagającego, stosownie do wskazań klinicznych.
Grupa farmakoterapeutyczna: leki immunosupresyjne, inne leki immunosupresyjne, kod ATC: L04AX07
Mechanizm działania
Mechanizm terapeutycznego działania fumaranu dimetylu w stwardnieniu rozsianym nie jest w pełni poznany. Wyniki badań przedklinicznych wskazują, że farmakodynamiczny efekt fumaranu dimetylu wynika głównie z aktywacji ścieżki transkrypcyjnej czynnika jądrowego Nrf2 [ ang. (erythroid derived 2) like 2]. Wykazano, że fumaran dimetylu zwiększa u pacjentów ekspresję genów ochrony antyoksydacyjnej zależnych od Nrf2 (takich jak np. dehydrogenaza NAD(P)H, chinon 1; [NQO1]).
Działanie farmakodynamiczne
Działanie na układ odpornościowy
W badaniach przedklinicznych i klinicznych wykazano właściwości przeciwzapalne i immunomodulacyjne fumaranu dimetylu. W modelach przedklinicznych fumaran dimetylu i fumaran monometylu, który jest głównym metabolitem fumaranu dimetylu, silnie hamowały aktywację komórek układu odpornościowego oraz wynikające z niej uwalnianie prozapalnych cytokin w odpowiedzi na bodźce zapalne. Co więcej, w badaniach klinicznych z udziałem pacjentów z łuszczycą fumaran dimetylu wpływał na fenotypy limfocytów poprzez zmniejszanie produkcji profilu cytokin prozapalnych (TH1, TH17) oraz pobudzał produkcję komórek przeciwzapalnych (TH2). Fumaran dimetylu wykazywał działanie terapeutyczne w wielorakich modelach urazów zapalnych i neurozapalnych. W badaniach III fazy u pacjentów ze stwardnieniem rozsianym (DEFINE, CONFIRM oraz ENDORSE) po rozpoczęciu leczenia produktem Tecfider0,9 × 109/l ), monitorowano pod kątem powrotu liczby limfocytów do DGN.
Rycina 1 przedstawia odsetek pacjentów, u których oszacowano metodą Kaplana-Meiera osiągnięcie DGN bez przedłużającej się ciężkiej limfopenii. Wartość wyjściową powrotu do normy (ang. recovery baseline, RBL) zdefiniowano jako ostatnią bezwzględną liczbę limfocytów (ang. Absolute Lymphocyte Count, ALC) w trakcie leczenia przed odstawieniem leczenia. Szacowany odsetek pacjentów, u których nastąpił powrót do wartości DGN (ALC ≥ 0,9 × 109 /l) w tygodniu 12. i 24., z łagodną, umiarkowaną lub ciężką limfopenią w punkcie RBL, przedstawiony jest w tabeli 1, tabeli 2 i tabeli 3, z 95% punktowymi przedziałami ufności. Błąd standardowy estymatora KaplanaMeiera w odniesieniu do funkcji przeżycia obliczano za pomocą wzoru Greenwoooda.
Ryc. 1: Metoda Kaplana-Meiera; odsetek pacjentów, u których nastąpił powrót do wartości ≥910 komórek/mm3 (0,9 × 109/l) DGN wobec wartości w punkcie wyjściowym RBL
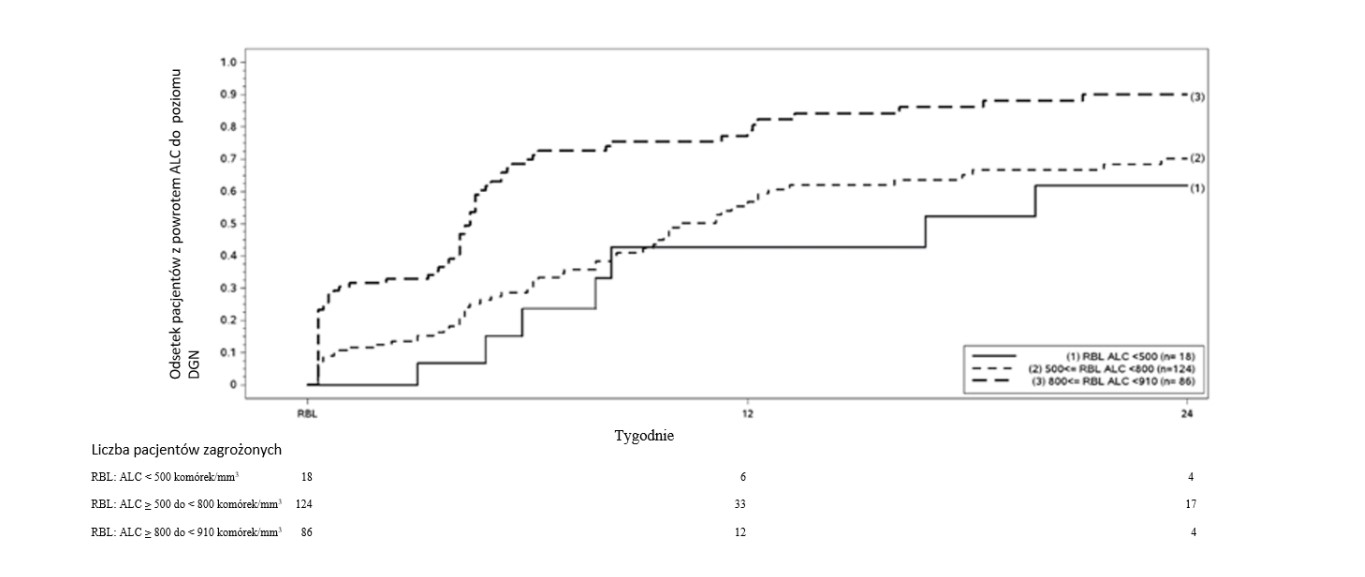
Uwaga: 500 komórek/mm3, 800 komórek/mm3, 910 komórek/mm3 odpowiada wartościom, odpowiednio, 0,5 × 109/l, 0,8 × 109/l i 0,9 × 109/l.
Tabela 1: Metoda Kaplana-Meiera; odsetek pacjentów, u których szacuje się osiągnięcie DGN, łagodna limfopenia w punkcie wyjściowym powrotu do normy (RBL), z wyłączeniem pacjentów z długotrwałą ciężką limfopenią
|
Liczba zagrożonych pacjentów z łagodną limfopeniąa
|
Okres wyjściowy N=86 |
Tydzień 12 N=12 |
Tydzień 24 N=4 |
|
Odsetek osiągających DGN (95% CI) |
|
0,81 (0,71; 0,89) |
0,90 (0,81; 0,96) |
a Pacjenci z ALC <0,9 × 109/l i >0,8 × 109/l w punkcie RBL, z wyłączeniem pacjentów z długotrwałą ciężką limfopenią.
Tabela 2: Metoda Kaplana-Meiera; odsetek pacjentów, u których szacuje się osiągnięcie DGN, umiarkowana limfopenia w punkcie wyjściowym powrotu do normy (RBL), z wyłączeniem pacjentów z długotrwałą ciężką limfopenią
|
Liczba zagrożonych pacjentów z umiarkowaną limfopeniąa
|
Okres wyjściowy N=124 |
Tydzień 12 N=33 |
Tydzień 24 N=17 |
|
Odsetek osiągających DGN (95% CI) |
|
0,57 (0,46; 0,67) |
0,70 (0,60; 0,80) |
aPacjenci z ALC <0,8 × 109/l i ≥0,5 × 109/l w punkcie RBL, z wyłączeniem pacjentów z długotrwałą ciężką limfopenią.
Tabela 3: Metoda Kaplana-Meiera; odsetek pacjentów, u których szacuje się osiągnięcie DGN, ciężka limfopenia w punkcie wyjściowym powrotu do normy (RBL), z wyłączeniem pacjentów z długotrwałą ciężką limfopenią
|
Liczba zagrożonych pacjentów z ciężką limfopeniąa
|
Okres wyjściowy N=18 |
Tydzień 12 N=6 |
Tydzień 24 N=4 |
|
Odsetek osiągających DGN (95% CI) |
|
0,43 (0,20; 0,75) |
0,62 (0,35; 0,88) |
aPacjenci z ALC <0,5 × 109/l w punkcie RBL, z wyłączeniem pacjentów z długotrwałą ciężką limfopenią.
Skuteczność kliniczna i bezpieczeństwo stosowania
Przeprowadzono dwa dwuletnie randomizowane badania kliniczne z zastosowaniem podwójnie ślepej próby, kontrolowane placebo (DEFINE z udziałem 1234 pacjentów i CONFIRM z udziałem 1417 pacjentów) z RRMS. W badaniach nie uczestniczyli pacjenci z postępującą postacią stwardnienia rozsianego.
Skuteczność (patrz tabela 4) i bezpieczeństwo wykazano u pacjentów z punktacją w skali EDSS (Rozszerzona Skala Niewydolności Ruchowej) od 0 do 5 włącznie, u których wystąpił co najmniej 1 rzut choroby w ciągu roku poprzedzającego włączenie do badania lub u których badanie mózgu MRI wykonane w ciągu 6 tygodni poprzedzających randomizację, wykazało co najmniej jedną zmianę Gd+ (po wzmocnieniu gadolinem). Badanie CONFIRM prowadzono metodą podwójnie ślepej próby (tj. badacz lub lekarz oceniający odpowiedź na badany lek też nie wie, który lek pacjent przyjmuje) z zastosowaniem leku porównawczego, octanu glatirameru.
Średnie wartości charakterystyki wyjściowej pacjentów w badaniu DEFINE przedstawiały się następująco: wiek 39 lat, czas trwania choroby 7,0 lat, punktacja w skali EDSS 2,0. Ponadto u 16% pacjentów wynik w skali EDSS wynosił >3,5, u 28% wystąpiły ≥2 rzuty w trakcie poprzedniego roku, a 42% było wcześniej leczonych innymi lekami stosowanymi zgodnie z zarejestrowanym wskazaniem w leczeniu stwardnienia rozsianego. W populacji pacjentów, u których wykonano badanie MRI, u 36% pacjentów włączonych do badania występowały w okresie wyjściowym zmiany Gd+ (średnia liczba zmian Gd+ wynosiła 1,4).
Średnie wartości charakterystyki wyjściowej pacjentów w badaniu CONFIRM przedstawiały się następująco: wiek 37 lat, czas trwania choroby 6,0 lat, punktacja w skali EDSS 2,5. Ponadto u 17% wynik w skali EDSS wynosił >3,5, u 32% wystąpiły ≥2 rzuty w trakcie poprzedniego roku, a 30% było wcześniej leczonych innymi lekami stosowanymi zgodnie z zarejestrowanym wskazaniem w leczeniu stwardnienia rozsianego. W populacji pacjentów, u których wykonano badanie MRI, u 45% pacjentów włączonych do badania występowały w okresie wyjściowym zmiany Gd+ (średnia liczba zmian Gd+ wynosiła 2,4).
W porównaniu do placebo u pacjentów leczonych produktem Tecfidera uzyskano klinicznie znaczącą i statystycznie istotną redukcję względem: pierwszorzędowego punktu końcowego w badaniu DEFINE, czyli odsetka pacjentów, u których wystąpił nawrót choroby po 2 latach, oraz pierwszorzędowego punktu końcowego w badaniu CONFIRM, czyli rocznego wskaźnika rzutów (nawrotów) (ang. annualized relapse rate, ARR) po 2 latach.
Tabela 4: Punkty końcowe kliniczne i MRI w badaniach DEFINE i CONFIRM
|
(DEFINE) |
(CONFIRM) |
||||
|
Placebo |
Tecfidera 240 mg |
Placebo |
Tecfidera 240 mg |
Octan glatiramer u |
|
|
Kliniczne punkty końcowea |
|||||
|
Liczba uczestników |
408 |
410 |
363 |
359 |
350 |
|
Roczny wskaźnik rzutów |
0,364 |
0,172*** |
0,401 |
0,224*** |
0,286* |
|
Częstość względna (95% CI–przedział ufności) |
0,47 (0,37; 0,61) |
0,56 (0,42; 0,74) |
0,71 (0,55; 0,93) |
||
|
Odsetek nawrotów |
0,461 |
0,270*** |
0,410 |
0,291** |
0,321** |
|
Ryzyko względne (95% CI–przedział ufności) |
0,51 (0,40; 0,66) |
0,66 (0,51; 0,86) |
0,71 (0,55; 0,92) |
||
|
Odsetek przypadków 12- tygodniowej potwierdzonej progresji |
0,271 |
0,164** |
0,169 |
0,128# |
0,156# |
|
Ryzyko względne (95% CI–przedział ufności) |
0,62 (0,44; 0,87) |
0,79 (0,52; 1,19) |
0,93 (0,63; 1,37) |
||
|
Odsetek przypadków 24- tygodniowej |
0,169 |
0,128# |
0,125 |
0,078# |
0,108# |
|
Ryzyko względne (95% CI–przedział ufności) |
0,77 (0,52; 1,14) |
0,62 (0,37; 1,03) |
0,87 (0,55; 1,38) |
||
|
Punkty końcowe MRIb |
|||||
|
Liczba uczestników |
165 |
152 |
144 |
147 |
161 |
|
Średnia (mediana) liczba nowych lub na nowo powiększających się zmian w obrazach |
16,5 (7,0) |
3,2 (1,0)*** |
19,9 (11,0) |
5,7 (2,0)*** |
9,6 (3,0)*** |
|
Średnia częstość zmian |
0,15 (0,10; 0,23) |
0,29 (0,21; 0,41) |
0,46 (0,33; 0,63) |
||
|
Średnia (mediana) liczba zmian Gd w ciągu 2 lat |
1,8 (0) |
0,1 (0)*** |
2,0 (0,0) |
0,5 (0,0)*** |
0,7 (0,0)** |
|
(DEFINE) |
(CONFIRM) |
||||
|
Placebo |
Tecfidera 240 mg dwa razy |
Placebo |
Tecfidera 240 mg dwa razy |
Octan glatiramer u |
|
|
Iloraz szans (95% CI–przedział ufności) |
0,10 (0,05; 0,22) |
0,26 (0,15; 0,46) |
0,39 (0,24; 0,65) |
||
|
Średnia (mediana) liczba nowych hipointensywnych zmian w obrazach T1-zależnych |
5,7 (2,0) |
2,0 (1,0)*** |
8,1 (4,0) |
3,8 (1,0)*** |
4,5 (2,0)** |
|
Średnia częstość zmian (95% CI–przedział ufności) |
0,28 (0,20; 0,39) |
0,43 (0,30; 0,61) |
0,59 (0,42; 0,82) |
||
aWszystkie analizy klinicznych punktów końcowych przeprowadzono na populacji ITT; b Do celów analizy wyników MRI wykorzystano kohortę badaną MRI
* P-wartość <0,05; ** P-wartość <0,01; *** P-wartość <0,0001; # statystycznie nieistotna
Do otwartego niekontrolowanego 8-letniego badania kontynuacyjnego (ENDORSE) włączono 1736 kwalifikujących się pacjentów z RRMS, którzy uczestniczyli w badaniach podstawowych (DEFINE i CONFIRM). Pierwszorzędowym celem badania była ocena długoterminowego bezpieczeństwa produktu Tecfidera u pacjentów z RRMS. Spośród 1736 pacjentów około połowa (909, 52%) była leczona przez 6 lat lub dłużej. We wszystkich 3 badaniach 501 pacjentów było stale leczonych produktem Tecfidera w dawce 240 mg dwa razy na dobę, a 249 pacjentów, którzy wcześniej otrzymywali placebo w badaniach DEFINE i CONFIRM, otrzymywało 240 mg dwa razy na dobę w badaniu ENDORSE. Pacjenci, którzy otrzymywali stałe leczenie dwa razy na dobę byli leczeni maksymalnie 12 lat.
Podczas badania ENDORSE ponad połowa wszystkich pacjentów leczonych produktem Tecfidera w dawce 240 mg dwa razy na dobę nie miała nawrotu choroby. W przypadku pacjentów stale leczonych dwa razy na dobę we wszystkich trzech badaniach, skorygowany wskaźnik ARR wynosił 0,187 (95% CI: 0,156; 0,224) w badaniach DEFINE i CONFIRM oraz 0,141 (95% CI: 0,119; 0,167) w badaniu ENDORSE. W przypadku pacjentów wcześniej leczonych placebo skorygowany wskaźnik ARR zmniejszył się z 0,330 (95% CI: 0,266; 0,408) w badaniach DEFINE i CONFIRM do 0,149 (95% CI: 0,116; 0,190) w badaniu ENDORSE.
W badaniu ENDORSE większość pacjentów (> 75%) nie miała potwierdzonej progresji niepełnosprawności (mierzonej jako progresja niepełnosprawności utrzymująca się przez 6 miesięcy). Połączone wyniki z trzech badań wykazały, że u pacjentów leczonych produktem Tecfidera odsetek przypadków potwierdzonej progresji niepełnosprawności był stały i niski, z niewielkim wzrostem średnich wyników w skali EDSS w całej populacji badania ENDORSE. Wyniki badania MRI (do 6. roku, obejmujące 752 pacjentów, którzy zostali wcześniej, w ramach badań DEFINE i CONFIRM, uwzględnieni w kohorcie, w której wykonywano badanie MRI) wykazały, że większość pacjentów (około 90%) nie miała zmian ulegających wzmocnieniu po podaniu gadolinu. W ciągu 6 lat skorygowana średnia liczba nowych lub nowo powiększających się zmian w obrazach T2-zależnych i nowych zmian w obrazach T1-zależnych pozostawała niska.
Skuteczność u pacjentów z silnie aktywną chorobą: W badaniach DEFINE i CONFIRM w podgrupie pacjentów z silnie aktywną chorobą obserwowano utrzymujące się działanie terapeutyczne wobec nawrotów, natomiast nie określono dokładnie skuteczności działania pod względem czasu do utrzymującej się przez 3 miesiące progresji niepełnosprawności ruchowej. Na potrzeby schematu badań, silnie aktywną chorobę zdefiniowano jak następuje:
- pacjenci z 2 lub więcej rzutami w ciągu jednego roku oraz z jedną lub więcej zmianami w obrazach mózgu po wzmocnieniu gadolinem (Gd+) w badaniu MRI (n=42 w badaniu DEFINE; n=51 w badaniu CONFIRM) lub
- pacjenci z brakiem odpowiedzi na pełne i odpowiednie leczenie (co najmniej rok leczenia) beta-interferonem; z co najmniej 1 rzutem w ciągu poprzedniego roku w trakcie leczenia, oraz z co najmniej 9 zmianami hiperintensywnymi w obrazach T2-zależnych w badaniu MRI mózgowia i co najmniej 1 zmianą Gd+, lub pacjenci z niezmienioną lub większą częstością rzutów w poprzednim roku w porównaniu do wcześniejszych 2 lat (n=177 w badaniu DEFINE; n=141 w badaniu CONFIRM).
Dzieci i młodzież
Bezpieczeństwo i skuteczność produktu Tecfidera u dzieci i młodzieży z RRMS oceniano w randomizowanym, otwartym badaniu prowadzonym z czynnym lekiem kontrolnym (interferon beta-1a) w grupach równoległych z udziałem pacjentów z RRMS w wieku od 10 do poniżej 18 lat. Stu pięćdziesięciu pacjentów przydzielono losowo do grupy przyjmującej fumaran dimetylu (240 mg dwa razy na dobę doustnie) lub do grupy stosującej interferon beta-1a (30 μg domięśniowo raz w tygodniu) przez 96 tygodni. Pierwszorzędowym punktem końcowym był odsetek pacjentów, u których w 96. tygodniu nie wystąpiły nowe lub nowo powiększające się zmiany hiperintensywne w obrazach T2- zależnych w badaniach mózgu metodą rezonansu magnetycznego. Przedstawiono statystyki opisowe, ponieważ nie zaplanowano wstępnie hipotezy potwierdzającej dla pierwszorzędowego punktu końcowego.
Odsetek pacjentów w populacji ITT bez nowych lub nowo powiększających się zmian w obrazach T2-zależnych w badaniu MRI w 96. tygodniu w stosunku do wartości wyjściowych wynosił 12,8% dla fumaranu dimetylu w porównaniu z 2,8% w grupie interferonu beta-1a. Średnia liczba nowych lub nowo powiększających się zmian w obrazach T2-zależnych w 96. tygodniu w stosunku do wartości wyjściowych, skorygowana o wyjściową liczbę zmian w obrazach T2-zależnych i wiek (populacja ITT z wykluczeniem pacjentów bez pomiarów MRI) wyniosła 12,4 dla fumaranu dimetylu i 32,6 dla interferonu beta-1a.
Prawdopodobieństwo klinicznego nawrotu wyniosło 34% w grupie fumaranu dimetylu i 48% w grupie interferonu beta-1a na koniec 96-tygodniowego okresu otwartego badania.
Profil bezpieczeństwa w zakresie oceny jakościowej u dzieci i młodzieży (w wieku od 13 do poniżej 18 lat) otrzymujących produkt Tecfidera był zgodny z wcześniej obserwowanym u pacjentów dorosłych (patrz punkt 4.8).
Podawany doustnie fumaran dimetylu podlega szybkiej przedukładowej hydrolizie przez esterazy i jest przekształcany do metabolitu pierwotnego, fumaranu monometylu, który jest również czynny. Fumaran dimetylu nie występuje w osoczu w mierzalnych stężeniach po doustnym podaniu produktu Tecfidera, a zatem wszystkie analizy farmakokinetyki dotyczące fumaranu dimetylu były przeprowadzane na podstawie osoczowych stężeń fumaranu monometylu. Dane farmakokinetyczne pochodziły od pacjentów ze stwardnieniem rozsianym oraz zdrowych ochotników.
Wchłanianie
Tmax fumaranu monometylu wynosi 2 do 2,5 godzin. Ponieważ kapsułki dojelitowe twarde Tecfidera zawierają mikrotabletki powlekane dojelitową powłoczką ochronną, wchłanianie rozpoczyna się dopiero, gdy opuszczą one żołądek (zwykle po upływie niecałej godziny). Po podaniu dawki 240 mg dwa razy dziennie z posiłkiem mediana maksymalnego stężenia (Cmax) wynosiła 1,72 mg/l, a całkowita ekspozycja wyrażona jako pole powierzchni pod krzywą (ang. area under the curve, AUC) wynosiła 8,02 mg·h/l u pacjentów ze stwardnieniem rozsianym. Łącznie Cmax i AUC zwiększały się w przybliżeniu proporcjonalnie do dawki w badanym zakresie dawek (120 mg do 360 mg). W badaniach z udziałem pacjentów ze stwardnieniem rozsianym dwie dawki po 240 mg podawano co 4 godziny w ramach schematu dawkowania TID (trzy razy dziennie). Wynikiem tego była minimalna akumulacja ekspozycji, dająca w rezultacie zwiększenie mediany Cmax o 12% w porównaniu do schematu dawkowania BID - dwa razy dziennie (1,72 mg/l w schemacie BID w porównaniu do 1,93 mg/l w schemacie TID) bez wpływu na bezpieczeństwo.
Pokarm nie wpływa w klinicznie istotnym stopniu na ekspozycję na fumaran dimetylu. Niemniej jednak produkt Tecfidera powinno się przyjmować z posiłkiem, gdyż poprawia to tolerancję odnośnie działań niepożądanych, takich jak nagłe zaczerwienienie skóry i dolegliwości żołądkowo-jelitowe (patrz punkt 4.2).
Dystrybucja
Pozorna objętość dystrybucji po doustnym podaniu 240 mg fumaranu dimetylu waha się pomiędzy 60 l a 90 l. U ludzi wiązanie fumaranu monometylu z białkami osocza na ogół waha się w przedziale od 27% do 40%.
Metabolizm
W organizmie ludzkim fumaran dimetylu jest w znacznym stopniu metabolizowany i mniej niż 0,1% dawki wydalana jest z moczem w postaci niezmienionej, jako fumaran dimetylu. Jest on wstępnie metabolizowany przez esterazy, obecne w całym przewodzie pokarmowym, krwi i tkankach, a następnie przedostaje się do krążenia układowego. Dalszy metabolizm odbywa się za pośrednictwem cyklu kwasów trikarboksylowych, bez udziału układu cytochromu P450 (CYP). W badaniu oceniającym pojedynczą dawkę 240 mg fumaranu dimetylu znakowanego węglem C14 jako główny metabolit w ludzkim osoczu zidentyfikowano glukozę. Do innych krążących metabolitów należały kwas fumarowy, kwas cytrynowy i fumaran monometylu. Metabolizm kolejnego produktu tego szlaku, kwasu fumarowego, odbywa się za pośrednictwem cyklu kwasów trikarboksylowych, przy czym główną drogą wydalania jest wydychanie w postaci dwutlenku węgla (CO2).
Eliminacja
Wydychanie CO2 to główna droga eliminacji fumaranu dimetylu, którą wydalane jest 60% dawki. Wydalanie z moczem i z kałem to wtórne drogi eliminacji, usuwające odpowiednio 15,5% i 0,9% dawki.
Okres półtrwania fumaranu monometylu w fazie eliminacji jest krótki (około 1 godziny) i po 24 godzinach w organizmie większości osób nie ma już pozostałości fumaranu monometylu. Po podawaniu wielokrotnych dawek fumaranu dimetylu w ramach schematu dawkowania nie następuje akumulacja fumaranu dimetylu ani też fumaranu monometylu.
Liniowość
Ekspozycja na fumaran dimetylu zwiększa się w przybliżeniu proporcjonalnie do dawki przy podawaniu pojedynczych i wielokrotnych dawek w badanym zakresie dawek, od 120 mg do 360 mg.
Farmakokinetyka u szczególnych grup pacjentów
W oparciu w wyniki analizy wariancji (ANOVA) masa ciała jest główną zmienną wpływającą na ekspozycję (wyrażoną w Cmax i AUC) u pacjentów z RRMS, ale czynnik ten nie wpływał na oceniane w badaniach klinicznych miary bezpieczeństwa i skuteczności.
Płeć i wiek nie wpływały w klinicznie istotnym stopniu na farmakokinetykę fumaranu dimetylu. Nie przeprowadzono badań farmakokinetyki u pacjentów w wieku 65 lat i starszych.
Dzieci i młodzież
Profil farmakokinetyczny fumaranu dimetylu podawanego w dawce 240 mg dwa razy na dobę został oceniony w niewielkim, otwartym niekontrolowanym badaniu z udziałem pacjentów w wieku od 13 do 17 lat (n=21) z RRMS. Farmakokinetyka produktu Tecfidera w tej grupie młodzieży była podobna do wcześniej obserwowanej u pacjentów dorosłych (Cmax: 2,00±1,29 mg/l; AUC0-12h: 3,62±1,16 h.mg/l, co odpowiada całkowitemu dziennemu AUC równemu 7,24 h.mg/l).
Zaburzenia czynności nerek
Z uwagi na fakt, że wydalanie przez nerki stanowi wtórną drogę eliminacji fumaranu dimetylu, którą usuwane jest mniej niż 16% podanej dawki, nie przeprowadzono oceny farmakokinetyki u osób z zaburzeniami nerek.
Zaburzenia czynności wątroby
Ponieważ fumaran dimetylu i fumaran monometylu są metabolizowane przez esterazy, bez udziału układu cytochromu P450 (CYP), nie przeprowadzono oceny farmakokinetyki u osób z zaburzeniami wątroby.
W badaniach klinicznych nie obserwowano niepożądanych działań opisanych poniżej w punktach „Toksykologia” oraz „Toksyczny wpływ na reprodukcję”, natomiast działania te obserwowano u zwierząt przy ekspozycji na poziomie podobnym do stężeń klinicznych.
Działanie genotoksyczne
Wyniki badań in vitro z zastosowaniem testu Amesa (test aberracji chromosomalnych w komórkach ssaków) były dla fumaranu dimetylu i fumaranu monometylu ujemne. Wyniki testu mikrojąderkowego in vivo u szczurów były dla fumaranu dimetylu ujemne.
Działanie rakotwórcze
Badania rakotwórczości fumaranu dimetylu prowadzono na myszach i szczurach przez okres do dwóch lat. Fumaran dimetylu podawano myszom w doustnych dawkach 25, 75, 200 i 400 mg/kg m.c./dobę oraz szczurom w dawkach 25, 50, 100 i 150 mg/kg m.c./dobę.
U myszy częstość występowania raka komórek kanalikowych nerek zwiększała się po dawce 75 mg/kg m.c./dobę, przy ekspozycji (AUC) odpowiadającej zalecanej dawce u ludzi. U szczurów częstość występowania raka komórek kanalikowych nerek i gruczolaków jąder z komórek Leydiga zwiększała się po dawce 100 mg/kg m.c./dobę – ekspozycji około 2 razy większej niż po zalecanej dawce u ludzi. Znaczenie wyników tych badań dla ryzyka u ludzi nie jest znane.
Częstość występowania brodawczaka płaskonabłonkowego i raka w przedżołądku (bezgruczołowej części żołądka) zwiększała się u myszy przy ekspozycji odpowiadającej dawce zalecanej dla ludzi, a u szczurów - przy ekspozycji poniżej tej dawki (w oparciu o AUC). U ludzi nie występuje odpowiednik przedżołądka gryzoni.
Toksykologia
Niekliniczne badania na gryzoniach, królikach i małpach prowadzono z zastosowaniem fumaranu dimetylu w postaci zawiesiny (fumaran dimetylu w 0,8% roztworze hydroksypropylometylocelulozy) podawanej drogą doustną przez zgłębnik. Badanie przewlekłej toksyczności u psów prowadzono z zastosowaniem podawanego doustnie fumaran dimetylu w postaci kapsułki.
Zmiany w nerkach obserwowano po wielokrotnym doustnym podawaniu fumaranu dimetylu u myszy, szczurów, psów i małp. Regenerację nabłonka kanalików nerkowych, sugerującą uszkodzenie, obserwowano u wszystkich gatunków. Rozrost kanalików nerkowych obserwowano u szczurów, którym produkt ten podawano przez całe życie (badanie dwuletnie). U psów, które przez 11 miesięcy otrzymywały codziennie doustną dawkę fumaranu dimetylu, wyliczona graniczna dawka, przy której stwierdzano zanik kory, była trzykrotnie większa niż dawka zalecana (w oparciu o AUC). U małp, które przez 12 miesięcy otrzymywały codziennie doustną dawkę fumaranu dimetylu, zaobserwowano martwicę pojedynczych komórek przy dawkach dwukrotnie większych niż dawka zalecana (w oparciu o AUC). Włóknienie śródmiąższowe i zanik kory obserwowano przy dawce sześciokrotnie większej niż dawka zalecana (w oparciu o AUC). Znaczenie wyników tych badań dla ryzyka u ludzi nie jest znane.
W jądrach szczurów i psów obserwowano zwyrodnienie nabłonka plemnikotwórczego. Efekty te obserwowano u szczurów po dawkach podobnych do dawki zalecanej, a u psów po dawkach trzykrotnie wyższych od dawki zalecanej (w oparciu AUC). Znaczenie wyników tych badań dla ryzyka u ludzi nie jest znane.
Zmiany zaobserwowane w przedżołądku myszy i szczurów w ramach badań trwających przez 3 miesiące i dłuższych obejmowały rozrost komórek płaskonabłonkowych (hiperplazję) i znaczne zgrubienie warstwy rogowej (hiperkeratozę); stany zapalne oraz występowanie brodawczaka płaskonabłonkowego i raka. U ludzi nie występuje odpowiednik przedżołądka gryzoni.
Toksyczny wpływ na reprodukcję i rozwój
Doustne podawanie fumaranu dimetylu samcom szczura w dawkach 75, 250 i 375 mg/kg m.c./dobę przed parzeniem i w okresie parzenia nie miało wpływu na płodność samców, aż do najwyższej badanej dawki (co najmniej dwa razy większej od dawki zalecanej na podstawie AUC). Doustne podawanie fumaranu dimetylu samicom szczura w dawkach 25, 100 i 250 mg/kg m.c./dobę przed parzeniem, w okresie parzenia i do 7. dnia ciąży, powodowało zmniejszenie liczby faz płodnych w okresie 14-dniowym oraz zwiększało liczbę zwierząt z przedłużonym okresem międzyrujowym po najwyższej badanej dawce (11 razy większej od dawki zalecanej na podstawie AUC). Zmiany te nie miały negatywnego wpływu na płodność ani na liczbę żywych płodów.
Wykazano, że u szczurów i królików fumaran dimetylu przenika przez błonę łożyskową do krwi płodu, przy czym stosunek stężenia w osoczu płodu do stężenia w osoczu matki wynosił odpowiednio 0,48- 0,64 i 0,1. Nie obserwowano wad rozwojowych po żadnej dawce fumaranu dimetylu u szczurów ani u królików. Podawanie fumaranu dimetylu w doustnych dawkach 25, 100 i 250 mg/kg m.c./dobę ciężarnym samicom szczura w okresie organogenezy wywoływało działania niepożądane u samic po dawkach czterokrotnie wyższych od dawki zalecanej na podstawie AUC oraz prowadziło do niskiej masy ciała płodów oraz opóźnienia kostnienia (w obrębie kości śródstopia i paliczków kończyn tylnych) po dawkach 11-krotnie wyższych od dawki zalecanej na podstawie AUC. Niższą masę ciała płodów i opóźnienie kostnienia uważano za efekt wtórny w stosunku do toksycznego działania na matkę (zmniejszenie masy ciała i ilości przyjmowanego pokarmu).
Doustne podawanie fumaranu dimetylu w dawkach 25, 75 i 150 mg/kg m.c./dobę ciężarnym samicom królika w okresie organogenezy nie wpływało w żaden sposób na rozwój zarodka i płodu, natomiast prowadziło do zmniejszenia masy ciała u matek po dawkach siedmiokrotnie wyższych od dawki zalecanej, a do większej liczby poronień po dawkach 16-krotnie wyższych od dawki zalecanej na podstawie AUC.
Doustne podawanie fumaranu dimetylu w dawkach 25, 100 i 250 mg/kg m.c./dobę samicom szczura w trakcie ciąży i laktacji prowadziło do obniżenia masy ciała u potomstwa w pokoleniu F1 oraz opóźnienia dojrzałości płciowej u samców pokolenia F1 po dawkach11-krotnie wyższych od dawki zalecanej na podstawie AUC. Nie wykazano wpływu na płodność potomstwa w pokoleniu F1. Niższą masę ciała potomstwa uważano za efekt wtórny w stosunku do toksycznego działania na matkę.
Toksyczność u młodych zwierząt
Dwa badania toksyczności u młodych szczurów, którym codziennie podawano doustnie fumaran dimetylu od 28. do 90-93. dnia po urodzeniu (co u ludzi odpowiada wiekowi około 3 lat i powyżej) wykazały podobną toksyczność wobec narządu docelowego (w nerkach i przedżołądku), jak u dorosłych zwierząt. W pierwszym badaniu fumaran dimetylu nie wpływał na rozwój, efekty neurobehawioralne ani płodność samców i samic aż do najwyższej dawki 140 mg/kg m.c./dobę (około 4,6-krotność zalecanej dawki u ludzi na podstawie ograniczonych danych dotyczących AUC u dzieci i młodzieży). Podobnie w drugim badaniu u młodych samców szczurów nie zaobserwowano wpływu na narządy rozrodcze i narządy dodatkowe samców aż do najwyższej dawki fumaranu dimetylu wynoszącej 375 mg/kg m.c./dobę (około 15-krotność przypuszczalnej wartości AUC przy zalecanej dawce dla dzieci i młodzieży). Zaobserwowano jednak zmniejszoną zawartość minerałów i gęstość kości w kości udowej i kręgach lędźwiowych u młodych samców szczurów. Zmiany w densytometrii kości obserwowano również u młodych szczurów po doustnym podaniu fumaranu diroksymelu, innego estru kwasu fumarowego, który jest metabolizowany do tego samego aktywnego metabolitu fumaranu monometylu in vivo. Poziom, przy którym nie obserwuje się działań niepożądanych (NOAEL) w zakresie zmian densytometrycznych u młodych szczurów wynosi około 1,5-krotności przypuszczalnej wartości AUC przy zalecanej dawce dla dzieci i młodzieży. Możliwy jest związek efektów kostnych z niższą masą ciała, ale nie można wykluczyć udziału efektu bezpośredniego. Wyniki dotyczące kości mają ograniczone znaczenie dla pacjentów dorosłych. W przypadku dzieci i młodzieży znaczenie tych wyników nie jest znane.
Zawartość kapsułki (mikrotabletki dojelitowe)
Celuloza mikrokrystaliczna
Kroskarmeloza sodowa
Talk
Koloidalny bezwodny krzemu dwutlenek
Magnezu stearynian
Trietylu cytrynian
Kwasu metakrylowego i metylu metakrylanu kopolimer (1:1)
Kwasu metakrylowego i etylu akrylanu kopolimer (1: 1) dyspersja 30%
Symetykon
Sodu laurylosiarczan Polisorbat 80
Otoczka kapsułki
Żelatyna
Tytanu dwutlenek (E171)
Błękit brylantowy FCF (E133)
Żelaza tlenek żółty (E172)
Nadruk kapsułki (czarny tusz)
Szelak
Potasu wodorotlenek
Żelaza tlenek czarny (E172)
Nie dotyczy.
4 lata
Nie przechowywać w temperaturze powyżej 30ºC.
Przechowywać blistry w opakowaniu zewnętrznym w celu ochrony przed światłem.
120 mg kapsułki dojelitowe, twarde: 14 kapsułek dojelitowych twardych w blistrach z PVC/PE/PVDC-PVC zamkniętych folią aluminiową.
240 mg kapsułki dojelitowe, twarde: 56 albo 168 kapsułek dojelitowych twardych w blistrach z PVC/PE/PVDC-PVC zamkniętych folią aluminiową.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Biogen Netherlands B.V.
Prins Mauritslaan 13
1171 LP Badhoevedorp
Holandia
EU/1/13/837/001
EU/1/13/837/002
EU/1/13/837/003
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 30 stycznia 2014
Data ostatniego przedłużenia pozwolenia: 15 września 2023
12/2024
Szczegółowe informacje o tym produkcie leczniczym są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu.
Powered by Biogen Poland Sp. z o.o.
(Biogen-101710)
 i
i  "Dodaj aplikację do ekranu początkowego"
"Dodaj aplikację do ekranu początkowego"