Powered by Biogen Poland Sp. z o.o.
Powered by Biogen Poland Sp. z o.o.
LEMTRADA 12 mg koncentrat do sporządzania roztworu do infuzji.
Każda fiolka zawiera 12 mg alemtuzumabu w 1,2 ml (10 mg/ml).
Alemtuzumab jest przeciwciałem monoklonalnym uzyskiwanym w zawiesinie zmodyfikowanych metodą rekombinacji DNA komórek ssaczych (komórki jajnika chomika chińskiego) hodowanych w podłożu odżywczym.
Substancje pomocnicze o znanym działaniu
Ten produkt leczniczy zawiera mniej niż 1 mmol potasu (39 mg) na wlew, to znaczy lek uznaje się za „wolny od potasu”.
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na wlew, to znaczy lek uznaje się za „wolny od sodu”.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Koncentrat do sporządzania roztworu do infuzji (koncentrat jałowy).
Przejrzysty, bezbarwny do lekko żółtego koncentrat o pH mieszczącym się w zakresie 7,0–7,4.
Produkt leczniczy LEMTRADA jest wskazany do stosowania w monoterapii do modyfikacji przebiegu choroby u dorosłych pacjentów z wysoce aktywną, rzutowo-ustępującą postacią stwardnienia rozsianego (ang. relapsing remitting multiple sclerosis, RRMS) w następujących grupach pacjentów:
lub
Leczenie produktem leczniczym LEMTRADA powinno być rozpoczynane i prowadzone wyłącznie pod nadzorem neurologa mającego doświadczenie w leczeniu pacjentów ze stwardnieniem rozsianym (SM), w warunkach szpitalnych z łatwym dostępem do intensywnej opieki medycznej. Należy zapewnić dostęp do specjalistów i sprzętu potrzebnego do przeprowadzenia w odpowiednim czasie diagnostyki i leczenia działań niepożądanych, zwłaszcza niedokrwienia mięśnia sercowego i zawału mięśnia sercowego, niepożądanych reakcji naczyniowo-mózgowych, chorób autoimmunologicznych i zakażeń.
Konieczne jest zabezpieczenie środków umożliwiających reagowanie na zespół uwalniania cytokin, reakcje nadwrażliwości i (lub) anafilaktyczne.
Pacjenci leczeni produktem leczniczym LEMTRADA muszą otrzymać Kartę ostrzegawczą pacjenta oraz Poradnik dla pacjenta. Ponadto należy ich poinformować o zagrożeniach związanych ze stosowaniem produktu leczniczego LEMTRADA (patrz również ulotka dla pacjenta).
Dawkowanie
Zalecana dawka alemtuzumabu to 12 mg na dobę podawane w postaci wlewu dożylnego w 2 początkowych cyklach leczenia z dodatkowymi dwoma cyklami leczenia, w razie potrzeby.
Początkowe dwa cykle leczenia:
W razie potrzeby można rozważyć do dwóch dodatkowych cyklów leczenia (patrz punkt 5.1):
Pominiętych dawek nie należy podawać tego samego dnia, w którym podawana jest dawka planowa.
Obserwacja pacjentów
Zaleca się terapię składającą się z 2 początkowych cyklów leczenia oraz, w razie potrzeby, do 2 dodatkowych cyklów leczenia (patrz dawkowanie) z zastosowaniem okresu monitorowania bezpieczeństwa pacjenta, trwającego od rozpoczęcia pierwszego cyklu leczenia przez co najmniej 48 miesięcy od ostatniego wlewu w ramach drugiego cyklu leczenia. Jeżeli zostanie zastosowany dodatkowy trzeci oraz czwarty cykl leczenia, okres monitorowania bezpieczeństwa pacjenta powinien być kontynuowany przez co najmniej 48 miesięcy od ostatniego wlewu (patrz punkt 4.4).
Premedykacja
Przez wszystkie 3 pierwsze dni każdego cyklu leczenia, bezpośrednio przed podaniem produktu leczniczego LEMTRADA, należy stosować u pacjentów premedykację kortykosteroidami. W badaniach klinicznych u pacjentów stosowano premedykację z użyciem 1000 mg metyloprednizolonu przez pierwsze 3 dni każdego cyklu leczenia produktem leczniczym LEMTRADA.
Przed podaniem produktu leczniczego LEMTRADA można również rozważyć zastosowanie premedykacji z użyciem leków przeciwhistaminowych i (lub) przeciwgorączkowych.
U wszystkich pacjentów, od pierwszego dnia każdego cyklu leczenia, należy wprowadzić doustną profilaktykę zakażenia wirusem herpes i kontynuować ją przez co najmniej 1 miesiąc po zakończeniu leczenia produktem leczniczym LEMTRADA (patrz również część „Zakażenia” w punkcie 4.4). W badaniach klinicznych pacjentom podawano acyklowir w dawce 200 mg dwa razy na dobę lub produkt równoważny.
Szczególne grupy pacjentów
Osoby w podeszłym wieku
W badaniach klinicznych nie uwzględniono pacjentów powyżej 61. roku życia. Nie określono, czy w ich wypadku odpowiedź na produkt leczniczy jest odmienna od tej obserwowanej u młodszych pacjentów.
Zaburzenia czynności nerek lub wątroby
Nie przeprowadzono badań dotyczących stosowania produktu leczniczego LEMTRADA u pacjentów z zaburzeniami czynności nerek lub wątroby.
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności produktu leczniczego LEMTRADA u dzieci w wieku od 0 do 18 lat chorujących na stwardnienie rozsiane. Nie powinno się stosować alemtuzumabu u dzieci w wieku od urodzenia do 10 roku życia do leczenia stwardnienia rozsianego. Dane nie są dostępne.
Sposób podawania
Przed wykonaniem wlewu konieczne jest rozcieńczenie produktu leczniczego LEMTRADA. Rozcieńczony roztwór należy podawać w postaci wlewu dożylnego trwającego około 4 godziny. Instrukcja dotycząca rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Pacjenci z zakażeniem ludzkim wirusem niedoboru odporności (HIV).
Pacjenci z ciężkim, czynnym zakażeniem, do czasu jego całkowitego ustąpienia.
Pacjenci z niekontrolowanym nadciśnieniem tętniczym.
Pacjenci z przebytym rozwarstwieniem tętnicy szyjnej i (lub) kręgowej.
Pacjenci z przebytym udarem mózgu. Pacjenci z przebytą dławicą piersiową lub zawałem mięśnia sercowego.
Pacjenci z rozpoznaną koagulopatią podczas leczenia przeciwpłytkowego lub przeciwzakrzepowego.
Pacjenci z innymi współistniejącymi chorobami autoimmunologicznymi, innymi niż stwardnienie rozsiane.
Produkt leczniczy LEMTRADA nie jest rekomendowany do leczenia pacjentów z nieaktywną postacią choroby ani tych, którzy są stabilni w trakcie aktualnego leczenia.
Pacjenci leczeni produktem leczniczym LEMTRADA muszą otrzymać Ulotkę dla pacjenta, Kartę ostrzegawczą pacjenta oraz Poradnik dla pacjenta. Przed rozpoczęciem leczenia, pacjentów należy poinformować o zagrożeniach i korzyściach związanych z leczeniem, a także o konieczności poddania się monitoringowi od momentu rozpoczęcia leczenia, aż do upływu co najmniej 48 miesięcy po ostatnim wlewie w ramach drugiego cyklu leczenia produktem leczniczym LEMTRADA. Jeżeli zostanie zastosowany dodatkowy cykl leczenia, monitorowanie bezpieczeństwa pacjenta powinno być kontynuowane aż do upływu co najmniej 48 miesięcy od ostatniego wlewu.
Identyfikowalność
W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Autoimmunizacja
Leczenie może skutkować wytworzeniem autoprzeciwciał i zwiększeniem ryzyka wystąpienia choroby autoimmunologicznej, która może być ciężka i zagrażająca życiu. Zgłaszano choroby autoimmunologiczne, w tym choroby tarczycy, immunologiczną plamicę małopłytkową (ang. immune thrombocytopenic purpura, ITP), nefropatie (np. chorobę z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych), autoimmunologiczne zapalenie wątroby (ang. autoimmune hepatitis, AIH), nabytą hemofilię A, zakrzepową plamicę małopłytkową (ang. thrombotic thrombocytopenic purpura, TTP), sarkoidozę oraz autoimmunologiczne zapalenie mózgu. Po wprowadzeniu produktu leczniczego LEMTRADA do obrotu, u leczonych nim pacjentów obserwowano wystąpienie licznych zaburzeń autoimmunologicznych. Pacjentów, u których rozwinie się autoimmunizacja, należy zbadać pod kątem innych chorób autoimmunologicznych (patrz punkt 4.3). Pacjenci i lekarze powinni zdawać sobie sprawę z możliwości wystąpienia zaburzeń autoimmunologicznych później niż po 48-miesięcznym okresie monitorowania.
Nabyta hemofilia A
Przypadki nabytej hemofilii A (przeciwciała przeciwko czynnikowi VIII) zgłaszano zarówno w badaniu klinicznym, jak i po wprowadzeniu do obrotu. U pacjentów pojawiają się zwykle samoistne krwiaki podskórne i rozległe siniaki, chociaż może także wystąpić krwiomocz, krwawienie z nosa, z przewodu pokarmowego lub inne rodzaje krwawień. U wszystkich pacjentów z takimi objawami należy wykonać koagulogram, uwzględniający czas kaolinowo-kefalinowy (ang. activated partial thromboplastin time, aPTT). W przypadku przedłużonego aPTT pacjent powinien zostać skierowany do lekarza hematologa. Należy poinformować pacjentów o objawach przedmiotowych i podmiotowych nabytej hemofilii A i konieczności natychmiastowego zwrócenia się po pomoc medyczną, w razie zaobserwowania któregokolwiek z tych objawów.
Zakrzepowa plamica małopłytkowa (TTP)
Po dopuszczeniu produktu do obrotu u pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano przypadki TTP, w tym zakończone zgonem. TTP to ciężki stan, który wymaga pilnej oceny i natychmiastowego leczenia, i może rozwinąć się dopiero kilka miesięcy po ostatnim wlewie produktu leczniczego LEMTRADA. TTP może charakteryzować się małopłytkowością, mikroangiopatyczną niedokrwistością hemolityczną, objawami neurologicznymi, gorączką i zaburzeniami czynności nerek.
Autoimmunologiczne zapalenie mózgu
U pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano przypadki autoimmunologicznego zapalenia mózgu. Autoimmunologiczne zapalenie mózgu charakteryzuje się podostrym początkiem (z szybkim postępem w ciągu miesięcy) zaburzeń pamięci, zmienionym stanem psychicznym lub objawami psychicznymi, zwykle w połączeniu z nowo pojawiającymi się ogniskowymi objawami neurologicznymi i napadami padaczkowymi. U pacjentów z podejrzeniem autoimmunologicznego zapalenia mózgu należy wykonać badanie neuroobrazowe (MRI), EEG, punkcję lędźwiową i testy serologiczne pod kątem odpowiednich biomarkerów (np. autoprzeciwciał przeciwneuronalnych) w celu potwierdzenia rozpoznania i wykluczenia innych etiologii.
Immunologiczna plamica małopłytkowa (ITP)
W ramach badań klinicznych prowadzonych z udziałem grupy kontrolnej dotyczących stwardnienia rozsianego u 12 (1%) leczonych pacjentów zaobserwowano ciężkie przypadki immunologicznej plamicy małopłytkowej (co odpowiada rocznemu wskaźnikowi 4,7 przypadków/1000 pacjentolat). Zaobserwowano też 12 kolejnych ciężkich przypadków immunologicznej plamicy małopłytkowej w ciągu (mediana) 6,1 lat obserwacji (maksymalnie 12 lat; skumulowany roczny wskaźnik: 2,8 przypadków/1000 pacjentolat). U jednego pacjenta doszło do rozwoju immunologicznej plamicy małopłytkowej, która nie została rozpoznana do momentu wykonania wymaganych comiesięcznych badań krwi i doprowadziła do zgonu pacjenta z powodu krwotoku śródmózgowego. W 79,5% przypadków immunologiczna plamica małopłytkowa ujawniała się w okresie 4 lat po pierwszej ekspozycji na produkt leczniczy. Jednak w niektórych przypadkach immunologiczna plamica małopłytkowa rozwijała się dopiero po latach. Do objawów immunologicznej plamicy małopłytkowej mogą należeć (choć nie tylko one): łatwe powstawanie zasinień, wybroczyny, samoistne krwawienie z błon śluzowych (np. krwawienie z nosa, krwioplucie), krwawienia miesiączkowe nieregularne lub o nasileniu większym niż zwykle. Krwioplucie może również wskazywać na chorobę z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych (patrz niżej) i wymaga przeprowadzenia odpowiedniego rozpoznania różnicowego. Należy przypominać pacjentowi o konieczności zachowania czujności pod kątem możliwych objawów oraz niezwłocznego zwrócenia się o pomoc medyczną w razie jakichkolwiek wątpliwości.
Przed rozpoczęciem leczenia, a następnie co miesiąc przez co najmniej 48 miesięcy od ostatniego wlewu, należy wykonywać badanie morfologiczne krwi z rozmazem. Po tym okresie należy przeprowadzić badania w oparciu o objawy kliniczne sugerujące obecność immunologicznej plamicy małopłytkowej. W razie podejrzenia wystąpienia immunologicznej plamicy małopłytkowej należy niezwłocznie wykonać badanie morfologiczne krwi.
W sytuacji potwierdzenia wystąpienia immunologicznej plamicy małopłytkowej należy natychmiast podjąć odpowiednią interwencję medyczną, w tym niezwłocznie skierować pacjenta do specjalisty. Dane pochodzące z badań klinicznych dotyczących stwardnienia rozsianego wykazały, że przestrzeganie zaleceń odnośnie monitorowania krwi i edukacja w zakresie objawów immunologicznej plamicy małopłytkowej umożliwiają wczesne jej wykrycie i rozpoczęcie leczenia, które w większości przypadków było skuteczne po zastosowaniu terapii pierwszej linii.
Nefropatie
Nefropatie, w tym chorobę z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych (choroba anty-GBM), zaobserwowano u 6 (0,4%) pacjentów biorących udział w badaniach klinicznych dotyczących stwardnienia rozsianego w okresie 6,1 lat obserwacji (mediana, maksymalnie 12 lat). Zazwyczaj pojawiały się one w ciągu 39 miesięcy po ostatnim podaniu produktu leczniczego LEMTRADA. W badaniach klinicznych stwierdzono 2 przypadki choroby z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych. Oba te przypadki miały ciężki przebieg, zostały wykryte wcześnie dzięki obserwacji klinicznej oraz monitorowaniu wyników badań laboratoryjnych i zakończyły się pomyślnie po zastosowaniu leczenia.
Do objawów klinicznych nefropatii mogą należeć zwiększenie stężenia kreatyniny w surowicy, krwiomocz i (lub) białkomocz. Chociaż nie zaobserwowano tego podczas badań klinicznych, skutkiem choroby z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych może być krwotok pęcherzykowy objawiający się krwiopluciem. Krwioplucie może również wskazywać na immunologiczną plamicę małopłytkową lub nabytą hemofilię A (patrz wyżej) i wymaga przeprowadzenia odpowiedniego rozpoznania różnicowego. Należy przypominać pacjentowi o zachowaniu czujności na możliwe objawy oraz o konieczności niezwłocznego zwrócenia się o pomoc medyczną w razie jakichkolwiek wątpliwości. Choroba z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych może prowadzić do niewydolności nerek wymagającej dializy i (lub) przeszczepu, jeśli szybko nie zostanie podjęte leczenie, i zagrażającej życiu w wypadku zaniechania leczenia.
Przed rozpoczęciem leczenia, a następnie co miesiąc przez co najmniej 48 miesięcy od ostatniego wlewu należy badać stężenie kreatyniny w surowicy. Przed rozpoczęciem leczenia, a następnie co miesiąc przez co najmniej 48 miesięcy od ostatniego wlewu, należy wykonywać mikroskopowe badanie osadu moczu. Stwierdzenie klinicznie istotnych zmian stężenia kreatyniny w surowicy w porównaniu z poziomem wyjściowym, wystąpienie niewyjaśnionego krwiomoczu i (lub) białkomoczu powinno skłonić do dalszej oceny w kierunku nefropatii, w tym do niezwłocznego skierowania do specjalisty. Wczesne rozpoznanie i rozpoczęcie leczenia nefropatii może zmniejszyć ryzyko niepomyślnych następstw. Po tym okresie należy przeprowadzać badania w oparciu o objawy kliniczne sugerujące obecność nefropatii.
Choroby tarczycy
W ramach badań klinicznych dotyczących stwardnienia rozsianego u około 36,8% pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg zaobserwowano zaburzenia układu dokrewnego związane z tarczycą, w tym autoimmunologiczne choroby tarczycy, w ciągu 6,1 lat (mediana) okresu obserwacji (maksymalnie 12 lat) po pierwszej ekspozycji na produkt leczniczy LEMTRADA. Zdarzenia związane z tarczycą występowały częściej u pacjentów z chorobami tarczycy w wywiadzie zarówno w grupie leczonej produktem leczniczym LEMTRADA, jak i w grupie leczonej interferonem beta 1a (IFNB-1a). Obserwowane autoimmunologiczne choroby tarczycy obejmowały nadczynność lub niedoczynność tego gruczołu. Większość zdarzeń miała nasilenie od łagodnego do umiarkowanego. Poważne zdarzenia wystąpiły u 4,4% pacjentów. Choroba Gravesa-Basedowa, nadczynność tarczycy i niedoczynność tarczycy, autoimmunologiczne zapalenie tarczycy oraz powiększenie tarczycy (wole) wystąpiły u więcej niż 1 pacjenta. Większość przypadków dotyczących gruczołu tarczowego leczono konwencjonalnymi metodami, jednak u niektórych pacjentów wymagana była interwencja chirurgiczna. Po wprowadzeniu do obrotu u kilku pacjentów, u których biopsja potwierdziła autoimmunologiczne zapalenie wątroby (ang. autoimmune hepatitis, AIH), wcześniej rozwinęły się autoimmunologiczne choroby tarczycy.
Przed rozpoczęciem leczenia, a następnie co 3 miesiące przez 48 miesięcy od ostatniego wlewu, należy wykonywać badania czynności tarczycy, takie jak oznaczenie stężenia tyreotropiny. Po tym czasie badania należy wykonywać w oparciu o dane kliniczne sugerujące nieprawidłową czynność tarczycy lub w przypadku ciąży.
Choroba tarczycy stanowi szczególne zagrożenie w przypadku kobiet w ciąży (patrz punkt 4.6).
W badaniach klinicznych zdarzenia związane z tarczycą wystąpiły u 74% pacjentów z dodatnim wynikiem testu na obecność przeciwciał przeciwko peroksydazie tarczycowej (anty-TPO) na początku badania oraz u 38% pacjentów z wynikiem ujemnym na początku badania. Zdecydowana większość (około 80%) pacjentów, u których wystąpiły zdarzenia związane z tarczycą po leczeniu, miała wyjściowo ujemny wynik testu na obecność przeciwciał anty-TPO. Wynika z tego, że niezależnie od wyniku testu na obecność przeciwciał anty-TPO przed rozpoczęciem leczenia, u pacjentów mogą wystąpić działania niepożądane związane z tarczycą. Należy zatem u wszystkich pacjentów okresowo wykonywać badania zgodnie z harmonogramem przedstawionym powyżej.
Cytopenie
W ramach badań klinicznych dotyczących stwardnienia rozsianego rzadko zgłaszano podejrzenie cytopenii autoimmunologicznych, takich jak neutropenia, niedokrwistość hemolityczna czy pancytopenia. W celu monitorowania cytopenii, w tym neutropenii, należy wykonywać badania morfologiczne krwi (patrz powyższy punkt poświęcony immunologicznej plamicy małopłytkowej). W razie potwierdzenia cytopenii należy niezwłocznie podjąć odpowiednią interwencję medyczną, w tym skierowanie do specjalisty.
Autoimmunologiczne zapalenie wątroby i uszkodzenie wątroby
U pacjentów leczonych produktem leczniczym LEMTRADA obserwowano przypadki autoimmunologicznego zapalenia wątroby (w tym również przypadki zgonów i przypadki wymagające przeszczepienia wątroby) oraz uszkodzenia wątroby związanego z zakażeniami (patrz punkt 4.3). Próby czynnościowe wątroby należy wykonywać przed rozpoczęciem leczenia i raz w miesiącu, co najmniej do 48 miesięcy po ostatnim dożylnym podaniu leku. Pacjenci powinni zostać poinformowani o ryzyku wystąpienia autoimmunologicznego zapalenia wątroby, uszkodzenia wątroby, a także o towarzyszących temu objawach.
Limfohistiocytoza hemofagocytarna (ang. haemophagocytic lymphohistiocytosis, HLH)
Po dopuszczeniu produktu do obrotu u pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano przypadki limfohistiocytozy hemofagocytarnej (w tym zakończone zgonem). HLH jest zagrażającym życiu zespołem patologicznej aktywacji immunologicznej, charakteryzującym się klinicznymi objawami skrajnie ciężkiego procesu zapalnego o charakterze ogólnoustrojowym. HLH cechuje gorączka, powiększenie wątroby i cytopenie. Stan ten wiąże się z wysokim ryzykiem zgonu, jeśli nie zostanie wcześnie rozpoznany i leczony. Występowanie objawów choroby zgłaszano w ciągu kilku miesięcy do czterech lat po rozpoczęciu leczenia. Pacjentów należy poinformować o objawach HLH i czasie, kiedy mogą wystąpić. Pacjentów, u których wystąpią wczesne objawy patologicznej aktywacji układu odpornościowego, należy niezwłocznie poddać ocenie i wziąć pod uwagę rozpoznanie HLH.
Reakcje związane z wlewem
W badaniach klinicznych reakcje związane z wlewem definiowano jako dowolne działanie niepożądane występujące w trakcie wlewu produktu leczniczego LEMTRADA lub w ciągu 24 godzin od jej zakończenia. Większość z tych reakcji może być spowodowana uwalnianiem cytokin w czasie wlewu. U większości pacjentów leczonych produktem leczniczym LEMTRADA w ramach badań klinicznych dotyczących stwardnienia rozsianego w czasie podawania produktu leczniczego LEMTRADA w dawce 12 mg i (lub) w ciągu 24 godzin po podaniu występowały reakcje związane z wlewem o nasileniu od łagodnego do umiarkowanego. Częstość reakcji związanych z wlewem była najwyższa podczas pierwszego cyklu leczenia. W trakcie całego okresu obserwacji, obejmującego też pacjentów otrzymujących dodatkowy cykl leczenia, do najczęstszych reakcji związanych z wlewem należały: ból głowy, wysypka, gorączka, nudności, pokrzywka, świąd, bezsenność, dreszcze, zaczerwienienie, zmęczenie, duszność, zaburzenia smaku, dyskomfort w klatce piersiowej, uogólniona wysypka, tachykardia, bradykardia, niestrawność, zawroty głowy i ból. Poważne reakcje wystąpiły u 3% pacjentów i obejmowały przypadki bólu głowy, gorączki, pokrzywki, tachykardii, migotania przedsionków, nudności, dyskomfortu w klatce piersiowej i niedociśnienia. Objawy kliniczne reakcji anafilaktycznych mogą wydawać się podobne do objawów klinicznych reakcji związanych z wlewem, jednak charakteryzują się wyższym stopniem ciężkości i mogą zagrażać życiu. Reakcje anafilaktyczne zgłaszano rzadziej niż reakcje związane z wlewem.
W celu złagodzenia reakcji związanych z wlewem zaleca się stosowanie premedykacji (patrz punkt 4.2).
W badaniach klinicznych z grupą kontrolną większość pacjentów otrzymała leki przeciwhistaminowe i (lub) przeciwgorączkowe przed co najmniej jednym wlewem produktu leczniczego LEMTRADA. Reakcje związane z wlewem mogą wystąpić u pacjentów niezależnie od wcześniejszego leczenia. Zaleca się obserwację pacjentów pod kątem reakcji związanych z wlewem w czasie podawania produktu leczniczego LEMTRADA oraz przez co najmniej 2 godziny po podaniu. W razie potrzeby należy rozważyć wydłużenie czasu obserwacji (hospitalizacji). W przypadku wystąpienia ciężkich reakcji związanych z wlewem, należy natychmiast przerwać wlew. Należy zapewnić odpowiednie środki na wypadek wystąpienia wstrząsu anafilaktycznego lub ciężkich reakcji (patrz poniżej).
Choroba Stilla u dorosłych (ang. Adult Onset Still’s disease, AOSD)
Po dopuszczeniu produktu do obrotu u pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano przypadki choroby Stilla u dorosłych (AOSD). Choroba ta jest rzadkim stanem zapalnym, który wymaga pilnej oceny i leczenia. U pacjentów z AOSD mogą wystąpić następujące objawy przedmiotowe lub podmiotowe: gorączka, zapalenie stawów, wysypka i leukocytoza przy braku infekcji, nowotwory złośliwe, a także inne choroby reumatyczne. Należy rozważyć przerwanie lub zaprzestanie leczenia produktem leczniczym LEMTRADA, jeżeli nie można ustalić alternatywnej etiologii tych objawów.
Inne ciężkie reakcje powiązane czasowo z wlewem produktu leczniczego LEMTRADA
Po dopuszczeniu produktu do obrotu zgłaszano rzadkie, ciężkie, niekiedy zakończone zgonem i nieprzewidziane działania niepożądane ze strony różnych układów narządów. W większości przypadków objawy występowały 1-3 dni po podaniu we wlewie produktu leczniczego LEMTRADA. Reakcje występowały po zastosowaniu którejkolwiek dawki leku, a także po 2. cyklu leczenia. Należy poinformować pacjentów o możliwych objawach oraz czasie ich wystąpienia. Należy poinformować pacjentów o możliwości opóźnionego wystąpienia reakcji związanych z wlewem dożylnym i poinstruować, aby zgłaszali objawy i szukali odpowiedniej opieki medycznej.
Udar krwotoczny
Wśród pacjentów, u których zgłoszono wystąpienie udaru, kilku było w wieku poniżej 50 lat i nie mieli oni w przeszłości nadciśnienia, zaburzeń krzepnięcia ani nie przyjmowali jednocześnie leków przeciwzakrzepowych lub przeciwpłytkowych. U niektórych pacjentów przed wystąpieniem krwotoku obserwowano podwyższoną wartość ciśnienia tętniczego krwi.
Niedokrwienie mięśnia sercowego i zawał mięśnia sercowego
Wśród pacjentów, u których zgłoszono wystąpienie niedokrwienia lub zawału, kilku było w wieku poniżej 40 lat i nie mieli oni czynników ryzyka choroby niedokrwiennej serca. Zauważono, że u niektórych pacjentów ciśnienie tętnicze krwi i (lub) częstość tętna były czasowo nieprawidłowe w trakcie wlewu dożylnego.
Rozwarstwienie tętnicy szyjnej i (lub) kręgowej
Przypadki rozwarstwienia tętnicy szyjnej i (lub) kręgowej, w tym rozwarstwienia wielokrotnego, zgłaszano zarówno w ciągu pierwszych dni po dożylnym podaniu produktu leczniczego LEMTRADA, jak i w późniejszym czasie, w ciągu pierwszego miesiąca po wlewie.
Krwawienie do światła pęcherzyków płucnychZgłoszone przypadki reakcji powiązanych czasowo z wlewem nie były związane z chorobą związaną z przeciwciałami przeciwko błonie podstawnej (anty-GBM; zespół Goodpasture’a).
Małopłytkowość
Zgłaszana mmałopłytkowość występowała w ciągu pierwszych dni po wlewie dożylnym (w przeciwieństwie do ITP). Często była samoograniczająca się i stosunkowo łagodna, chociaż w wielu przypadkach jej nasilenie i zakończenie były nieznane.
Zapalenie osierdzia
Zgłaszano rzadkie przypadki zapalenia osierdzia, wysięku osierdziowego i innych zdarzeń dotyczących osierdzia, zarówno w ramach ostrej, jak i później reakcji na wlew.
Zapalenie płuc
U pacjentów po podaniu wlewu produktu leczniczego LEMTRADA zgłaszano przypadki zapalenia płuc. Większość przypadków wystąpiła w pierwszym miesiącu po rozpoczęciu leczenia produktem leczniczym LEMTRADA. Pacjentom należy zalecić zgłaszanie lekarzowi objawów zapalenia płuc, które mogą obejmować skrócenie oddechu (duszność), kaszel, świszczący oddech, ból lub ucisk w klatce piersiowej i krwioplucie.
Instrukcje dotyczące wlewu w celu zmniejszenia występowania ciężkich reakcji niedługo po podaniu wlewu dożylnego produktu leczniczego Lemtrada
Zakażenia
W trwających przez maksymalnie 2 lata badaniach klinicznych z grupą kontrolną dotyczących stwardnienia rozsianego, zakażenia wystąpiły u 71% pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg i u 53% pacjentów leczonych podawanym podskórnie interferonem beta-1a [IFNB 1a] (44 mikrogramy 3 razy w tygodniu). Zakażenia te miały przeważnie nasilenie od łagodnego do umiarkowanego. Zakażenia, które występowały częściej u pacjentów leczonych produktem leczniczym LEMTRADA niż u pacjentów leczonych IFNB 1a, obejmowały zapalenie nosogardzieli, zakażenia układu moczowego, zakażenia górnych dróg oddechowych, zapalenie zatok, opryszczkowe zapalenie jamy ustnej, grypę i zapalenie oskrzeli. W ramach badań klinicznych z udziałem grupy kontrolnej dotyczących stwardnienia rozsianego ciężkie zakażenia wystąpiły u 2,7% pacjentów leczonych produktem leczniczym LEMTRADA i u 1% pacjentów leczonych IFNB-1a. Zakażenia o ciężkim przebiegu stwierdzone w grupie leczonej produktem leczniczym LEMTRADA obejmowały: zapalenie wyrostka robaczkowego, zapalenie żołądka i jelit, zapalenie płuc, półpasiec i zakażenia w obrębie zębów. Czas trwania zakażeń był zazwyczaj standardowy, a objawy ustępowały po zastosowaniu konwencjonalnego leczenia.
Skumulowany roczny wskaźnik zakażeń wyniósł 0,99 w ciągu 6,1 lat obserwacji (mediana) (maksymalnie 12 lat) po pierwszej ekspozycji na produkt leczniczy LEMTRADA, w porównaniu z 1,27 w badaniach klinicznych z grupą kontrolną.
Ciężkie zakażenia wirusem ospy wietrznej i półpaśca, obejmujące zakażenie pierwotne i reaktywację wirusa, występowały podczas badań klinicznych częściej wśród pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg (0,4%) niż u pacjentów przyjmujących IFNB-1a (0%). U 2% pacjentek leczonych produktem leczniczym LEMTRADA w dawce 12 mg zgłaszano również zakażenia wirusem brodawczaka ludzkiego (HPV) w obrębie szyjki macicy, w tym dysplazję szyjki macicy i kłykciny kończyste (brodawki okolicy odbytowej). W przypadku pacjentek zalecane jest przeprowadzanie raz w roku badania przesiewowego pod kątem zakażenia wirusem HPV.
Wśród osób leczonych produktem leczniczym LEMTRADA zgłaszano zakażenia wirusem cytomegalii (CMV), w tym przypadki reaktywacji wirusa. Większość z nich wystąpiła w ciągu 2 miesięcy od rozpoczęcia leczenia. Przed rozpoczęciem leczenia można rozważyć ocenę statusu serologicznego pacjenta zgodnie z miejscowymi wytycznymi.
U pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano zakażenie wirusem Epsteina-Barr (EBV), w tym reaktywację oraz ciężkie, a czasami śmiertelne, przypadki zapalenia wątroby wywołanego przez wirusa EBV.
Podczas badań klinicznych z udziałem grupy kontrolnej u pacjentów leczonych produktem leczniczym LEMTRADA oraz IFNB-1a zgłaszano występowanie gruźlicy. Czynną i utajoną gruźlicę, w tym kilka przypadków gruźlicy rozsianej, stwierdzono u 0,3% pacjentów leczonych produktem leczniczym LEMTRADA, częściej na obszarach endemicznych. Przed rozpoczęciem leczenia u wszystkich pacjentów należy przeprowadzić badania pod kątem zarówno aktywnego, jak i nieaktywnego (utajonego) zakażenia gruźliczego, zgodnie z lokalnymi wytycznymi.
U pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano występowanie listeriozy (zapalenie opon mózgowych wywołane przez bakterie z rodzaju Listeria), zwykle w ciągu miesiąca po wlewie produktu leczniczego LEMTRADA. Aby ograniczyć ryzyko zakażenia, pacjenci otrzymujący produkt leczniczy LEMTRADA powinni unikać spożywania surowych lub niedopieczonych mięs, serów pleśniowych oraz niepasteryzowanych produktów mlecznych przez dwa tygodnie przed wlewem, w trakcie oraz co najmniej miesiąc po wlewie produktu leczniczego LEMTRADA.
W badaniach klinicznych z grupą kontrolną dotyczących stwardnienia rozsianego powierzchowne zakażenia grzybicze, w szczególności kandydoza jamy ustnej i pochwy, częściej występowały u pacjentów leczonych produktem leczniczym LEMTRADA (12%) niż u pacjentów otrzymujących IFNB-1a (3%).
U pacjentów z ciężkim, czynnym zakażeniem, rozpoczęcie leczenia produktem leczniczym LEMTRADA należy opóźnić do czasu ustąpienia zakażenia. Pacjentów otrzymujących produkt leczniczy LEMTRADA należy poinstruować, aby zgłaszali objawy zakażeń lekarzowi.
Profilaktykę z zastosowaniem doustnych leków przeciw wirusowi herpes należy wprowadzić od pierwszego dnia leczenia produktem leczniczym LEMTRADA i kontynuować ją przez co najmniej 1 miesiąc po zakończeniu każdego cyklu leczenia. W badaniach klinicznych pacjentom podawano acyklowir w dawce 200 mg dwa razy na dobę lub produkt równoważny.
Produkt leczniczy LEMTRADA nie był podawany w celu leczenia stwardnienia rozsianego jednocześnie z produktami przeciwnowotworowymi i immunosupresyjnymi ani po podaniu tych produktów. Tak jak w przypadku innych terapii immunomodulacyjnych, rozważając podanie produktu leczniczego LEMTRADA, należy wziąć pod uwagę możliwy złożony wpływ produktów leczniczych na układ immunologiczny pacjenta. Jednoczesne stosowanie produktu leczniczego LEMTRADA z każdym z tych produktów może zwiększać ryzyko rozwoju immunosupresji.
Brak danych dotyczących powiązania produktu leczniczego LEMTRADA z reaktywacją wirusa zapalenia wątroby typu B (WZW B) lub zapalenia wątroby typu C (WZW C), gdyż z udziału w badaniach klinicznych wykluczano pacjentów z objawami czynnego lub przewlekłego zakażenia. Należy rozważyć wykonanie przed rozpoczęciem podawania produktu leczniczego LEMTRADA badania przesiewowego u pacjentów z wysokim ryzykiem zakażenia wirusem WZW B i (lub) WZW C, a także należy zachować ostrożność przepisując produkt leczniczy LEMTRADA pacjentom będącym nosicielami wirusa WZW B i (lub) WZW C, gdyż w ich przypadku może istnieć ryzyko nieodwracalnego uszkodzenia wątroby na skutek potencjalnej reaktywacji wirusa wynikającej z ich wcześniejszego stanu zdrowia.
Postępująca wieloogniskowa leukoencefalopatia (ang. Progressive Multifocal Leukoencephalopathy, PML)
U pacjentów z SM, po leczeniu alemtuzumabem, zgłaszano rzadkie przypadki (w tym śmiertelne) postępującej wieloogniskowej leukoencefalopatii (PML). Pacjentów leczonych alemtuzumabem należy monitorować w celu wykrycia objawów wskazujących na PML. Czynniki ryzyka o szczególnym znaczeniu obejmują wcześniejsze leczenie immunosupresyjne, zwłaszcza inne terapie stosowane w leczeniu SM, o których wiadomo, że wiążą się z ryzykiem wywoływania PML.
Zmiany w badaniu MRI mogą być widoczne przed ujawnieniem się objawów klinicznych. Przed rozpoczęciem i ponownym podaniem leczenia alemtuzumabem należy wykonać badanie MRI i ocenić pod kątem objawów związanych z PML. W razie potrzeby należy przeprowadzić dalszą diagnostykę, w tym badanie płynu mózgowo-rdzeniowego (ang. cerebrospinal fluid, CSF) na obecność DNA wirusa JC i powtórne badanie neurologiczne. Lekarz powinien zwrócić szczególną uwagę na objawy sugerujące PML, których pacjent może nie zauważyć (np. zaburzenia poznawcze, neurologiczne lub psychiczne). Pacjentom należy również doradzić, aby poinformowali o swoim leczeniu krewnych lub opiekunów, ponieważ mogą oni zauważyć objawy, których pacjent nie jest świadomy. PML należy uwzględnić w diagnostyce różnicowej u każdego pacjenta z SM przyjmującego alemtuzumab z objawami neurologicznymi i (lub) nowymi zmianami w mózgu w badaniu MRI.
Jeśli rozpoznano PML, nie należy rozpoczynać ani wznawiać leczenia alemtuzumabem.
Ostre niekamicze zapalenie pęcherzyka żółciowego
Produkt leczniczy LEMTRADA może zwiększać ryzyko wystąpienia ostrego niekamiczego zapalenia pęcherzyka żółciowego. W kontrolowanych badaniach klinicznych ostre niekamicze zapalenie pęcherzyka żółciowego występowało u 0,2% pacjentów ze stwardnieniem rozsianym leczonych produktem leczniczym LEMTRADA, w porównaniu z 0% pacjentów leczonych IFNB-1a. W okresie po wprowadzeniu produktu leczniczego do obrotu, u pacjentów leczonych produktem leczniczym LEMTRADA zgłaszano dodatkowe przypadki ostrego niekamiczego zapalenia pęcherzyka żółciowego. Czas do wystąpienia objawów po wlewie produktu leczniczego LEMTRADA wynosił od mniej niż 24 godziny do 2 miesięcy. Większość pacjentów leczono zachowawczo antybiotykami i wyzdrowiała bez interwencji chirurgicznej, inni zostali poddani zabiegowi cholecystektomii. Objawy ostrego niekamiczego zapalenia pęcherzyka żółciowego obejmują ból brzucha, tkliwość brzucha, gorączkę, nudności i wymioty. Ostre niekamicze zapalenie pęcherzyka żółciowego, niezdiagnozowane odpowiednio wcześnie i nieleczone, może być związane z wysokimi wskaźnikami zachorowalności i umieralności. W razie podejrzenia wystąpienia ostrego niekamiczego zapalenia pęcherzyka żółciowego, należy postawić diagnozę i niezwłocznie podjąć leczenie.
Nowotwory złośliwe
Tak jak w przypadku innych terapii immunomodulacyjnych, rozpoczynając podanie produktu leczniczego LEMTRADA, należy zachować ostrożność u pacjentów z występującym wcześniej i (lub) aktywnym nowotworem złośliwym. Obecnie nie wiadomo, czy produkt leczniczy LEMTRADA zwiększa ryzyko wystąpienia nowotworów złośliwych tarczycy, jednak sama tylko autoimmunologiczna choroba tarczycy może być czynnikiem ryzyka nowotworów złośliwych tarczycy.
Antykoncepcja
U myszy w czasie ciąży i po porodzie obserwowano przenikanie przez łożysko produktu leczniczego LEMTRADA i jego potencjalną aktywność farmakologiczną. Kobiety w wieku rozrodczym powinny w czasie leczenia i przez 4 miesiące po zakończeniu cyklu leczenia produktem leczniczym LEMTRADA stosować skuteczną metodę antykoncepcji (patrz punkt 4.6).
Szczepionki
Zaleca się, aby pacjenci spełnili lokalne wymagania dotyczące szczepień na co najmniej 6 tygodni przed rozpoczęciem leczenia produktem leczniczym LEMTRADA. Nie badano możliwości wytworzenia odpowiedzi immunologicznej na jakąkolwiek szczepionkę po zakończeniu leczenia produktem leczniczym LEMTRADA.
Po cyklu leczenia produktem leczniczym LEMTRADA nie przeprowadzono oficjalnych badań klinicznych z grupą kontrolną dotyczących bezpieczeństwa immunizacji pacjentów chorujących na stwardnienie rozsiane z użyciem szczepionek zawierających żywe wirusy, w związku z czym nie należy podawać tego typu szczepionek pacjentom ze stwardnieniem rozsianym, którzy niedawno przeszli taki cykl leczenia.
Wykrywanie przeciwciał przeciw wirusowi ospy wietrznej i półpaśca/szczepienie
Podobnie jak w przypadku każdego produktu leczniczego o działaniu immunomodulacyjnym, przed rozpoczęciem cyklu leczenia produktem leczniczym LEMTRADA pacjenci, którzy nie chorowali na ospę wietrzną lub którzy nie zostali zaszczepieni przeciwko wirusowi ospy wietrznej i półpaśca (VZV), powinni zostać przebadani pod kątem obecności przeciwciał przeciw wirusowi VZV. U pacjentów, u których nie wykryto tych przeciwciał, należy rozważyć wykonanie szczepienia przeciw wirusowi VZV przed rozpoczęciem leczenia produktem leczniczym LEMTRADA. Aby umożliwić wystąpienie pełnego działania szczepionki przeciw wirusowi VZV, należy odłożyć leczenie produktem leczniczym LEMTRADA na okres 6 tygodni po szczepieniu.
Zalecane badania laboratoryjne w celu monitorowania pacjentów
Przez co najmniej 48 miesięcy po ostatnim cyklu leczenia produktem leczniczym LEMTRADA należy okresowo wykonywać badania lekarskie i badania laboratoryjne w celu monitorowania pacjentów pod kątem wczesnych objawów choroby autoimmunologicznej:
Informacje dotyczące stosowania alemtuzumabu uzyskane przed wprowadzeniem produktu leczniczego LEMTRADA do obrotu, pochodzące ze źródeł innych niż badania sponsorowane przez firmę
Poniżej przedstawiono działania niepożądane zidentyfikowane przed rejestracją produktu leczniczego LEMTRADA, zgłaszane podczas stosowania alemtuzumabu w leczeniu przewlekłej białaczki limfocytowej B-komórkowej (PBL-B), jak również innych chorób, na ogół z wykorzystaniem wyższych i częściej podawanych dawek (np. 30 mg) niż zalecana w leczeniu stwardnienia rozsianego. Działania te były zgłaszane dobrowolnie przez pacjentów z populacji o zmiennej wielkości, dlatego nie zawsze było możliwe wiarygodne oszacowanie częstości ich występowania lub związku przyczynowego z ekspozycją na alemtuzumab.
Choroba autoimmunologiczna
Do zdarzeń autoimmunologicznych zgłaszanych przez pacjentów leczonych alemtuzumabem należały: neutropenia, niedokrwistość hemolityczna (w tym przypadek śmiertelny), nabyta hemofilia, choroba z obecnością przeciwciał przeciwko błonie podstawnej kłębuszków nerkowych i choroba tarczycy. U pacjentów leczonych alemtuzumabem, którzy nie chorowali na stwardnienie rozsiane, stwierdzono ciężkie i czasem śmiertelne zdarzenia autoimmunologiczne, w tym autoimmunologiczną niedokrwistość hemolityczną, autoimmunologiczną małopłytkowość, niedokrwistość aplastyczną, zespół Guillaina-Barrégo oraz przewlekłą zapalną poliradikuloneuropatię demielinizacyjną. U pacjenta onkologicznego leczonego alemtuzumabem zgłoszono dodatni odczyn Coombsa. Inny pacjent onkologiczny leczony alemtuzumabem zmarł w wyniku choroby przeszczep przeciw gospodarzowi związanej z przetoczeniem krwi.
Reakcje związane z wlewem
U pacjentów, którzy nie chorowali na stwardnienie rozsiane i byli leczeni alemtuzumabem z użyciem większych i częściej podawanych dawek niż w przypadku leczenia stwardnienia rozsianego, występowały ciężkie i czasem śmiertelne reakcje związane z wlewem, w tym skurcz oskrzeli, hipoksja, utrata przytomności, nacieki płucne, zespół ostrej niewydolności oddechowej, zatrzymanie oddechu, zawał mięśnia sercowego, zaburzenia rytmu serca, ostra niewydolność serca i zatrzymanie akcji serca. Zgłaszano również ciężkie reakcje anafilaktyczne i inne reakcje nadwrażliwości, w tym wstrząs anafilaktyczny i obrzęk naczynioruchowy.
Zakażenia i zarażenia pasożytnicze
U pacjentów, którzy nie chorowali na stwardnienie rozsiane i byli leczeni alemtuzumabem z użyciem większych i częściej podawanych dawek niż w przypadku leczenia stwardnienia rozsianego, występowały ciężkie i czasem śmiertelne zakażenia wirusowe, bakteryjne, pierwotniakowe i grzybicze, w tym wynikające z reaktywacji utajonych zakażeń.
Zaburzenia krwi i układu chłonnego
U pacjentów, którzy nie chorowali na stwardnienie rozsiane, zgłaszano przypadki ciężkich krwawień.
Zaburzenia serca
U pacjentów leczonych alemtuzumabem, którzy nie chorowali na stwardnienie rozsiane i którym podawano wcześniej środki o potencjalnym działaniu kardiotoksycznym, zgłaszano występowanie zastoinowej niewydolności serca, kardiomiopatii oraz zmniejszenie frakcji wyrzutowej.
Zaburzenia limfoproliferacyjne związane z wirusem Epsteina-Barr
W ramach badań sponsorowanych przez inne firmy obserwowano zaburzenia limfoproliferacyjne związane z zakażeniem wirusem Epsteina-Barr
Produkt leczniczy LEMTRADA zawiera sód i potas
Ten produkt leczniczy zawiera mniej niż 1 mmol potasu (39 mg) na wlew, to znaczy lek uznaje się za „wolny od potasu”.
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na wlew, to znaczy lek uznaje się za „wolny od sodu”.
Nie przeprowadzono oficjalnych badań dotyczących interakcji produktu leczniczego LEMTRADA stosowanego w zalecanej dawce z innymi lekami u pacjentów cierpiących na stwardnienie rozsiane. W kontrolowanych badaniach klinicznych dotyczących pacjentów chorujących na stwardnienie rozsiane i leczonych ostatnio interferonem beta i octanem glatirameru, wymagane było przerwanie leczenia na 28 dni przed rozpoczęciem podawania produktu leczniczego LEMTRADA.
Kobiety w wieku rozrodczym
Stężenia w surowicy były niskie lub niewykrywalne po około 30 dniach od zakończenia każdego cyklu leczenia. Z tego powodu podczas każdego cyklu leczenia produktem leczniczym LEMTRADA i do 4 miesięcy po jego zakończeniu kobiety w wieku rozrodczym muszą stosować skuteczną antykoncepcję.
Ciąża
Istnieją ograniczone dane dotyczące stosowania alemtuzumabu u kobiet w okresie ciąży. Produkt leczniczy LEMTRADA może być podawany w czasie ciąży jedynie, jeśli potencjalne korzyści przewyższają potencjalne zagrożenie dla płodu.
Ludzka immunoglobulina G (IgG) przenika przez barierę łożyska; alemtuzumab również może przenikać przez łożysko i w związku z tym stwarzać zagrożenie dla płodu. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Nie wiadomo, czy alemtuzumab podawany kobietom w okresie ciąży może wpływać na płodność kobiet oraz doprowadzić do uszkodzeń płodu.
Choroba tarczycy (patrz punkt 4.4 Choroby tarczycy) stanowi szczególne zagrożenie w przypadku kobiet w ciąży. W razie braku leczenia niedoczynności tarczycy w trakcie ciąży istnieje zwiększone ryzyko poronienia i negatywnego wpływu na płód, obejmującego opóźnienie umysłowe i karłowatość. U kobiet z chorobą Gravesa-Basedowa matczyne przeciwciała przeciw tyreotropinie mogą zostać przekazane rozwijającemu się płodowi, co może prowadzić do rozwoju przejściowej noworodkowej postaci choroby Gravesa-Basedowa.
Karmienie piersią
Alemtuzumab był obecny w mleku oraz w organizmie potomstwa samic myszy w okresie laktacji.
Nie wiadomo, czy alemtuzumab przenika do mleka kobiecego. Nie można wykluczyć zagrożenia dla noworodków/niemowląt karmionych piersią. Z tego powodu należy przerwać karmienie piersią na czas trwania każdego cyklu leczenia produktem leczniczym LEMTRADA oraz na 4 miesiące po ostatnim wlewie w każdym cyklu. Jednak korzyści wynikające z nabycia odporności wraz z mlekiem matki mogą przeważać nad ryzykiem potencjalnej ekspozycji na alemtuzumab u noworodków/niemowląt karmionych piersią.
Płodność
Brak odpowiednich danych klinicznych dotyczących bezpieczeństwa na temat wpływu produktu leczniczego LEMTRADA na płodność. W badaniu cząstkowym przeprowadzonym u 13 mężczyzn leczonych produktem leczniczym LEMTRADA (w dawce 12 mg lub 24 mg) nie stwierdzono aspermii, azoospermii, stale obniżonej liczby plemników, zaburzeń motoryki czy zwiększenia nieprawidłowości w budowie morfologicznej plemników.
Wiadomo, że antygen CD52 jest obecny na powierzchni komórek układu rozrodczego ludzi i gryzoni. Badania na zwierzętach wykazały wpływ na płodność u humanizowanych myszy (patrz punkt 5.3), jednak na podstawie dostępnych danych potencjalny wpływ na płodność u ludzi w czasie ekspozycji nie jest znany.
Produkt leczniczy LEMTRADA ma mały wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U większości pacjentów wystąpiły reakcje związane z wlewem podczas podawania lub w ciągu 24 godzin od podania produktu leczniczego LEMTRADA. Niektóre z reakcji związanych z wlewem (np. zawroty głowy) mogą przejściowo zaburzać zdolność pacjenta do prowadzenia pojazdów i obsługiwania maszyn i należy zachować ostrożność, aż do ich ustąpienia.
Podsumowanie profilu bezpieczeństwa w badaniach klinicznych
Populacja objęta oceną bezpieczeństwa w łącznej analizie danych pochodzących z badań klinicznych nad stwardnieniem rozsianym liczyła 1486 pacjentów leczonych produktem leczniczym LEMTRADA (w dawce 12 mg lub 24 mg). Z medianą okresu obserwacji wynoszącą 6,1 lat (maksymalnie 12 lat) dało to 8635 pacjentolat okresu obserwacji.
Najważniejszymi działaniami niepożądanymi były choroby autoimmunologiczne (immunologiczna plamica małopłytkowa, choroby tarczycy, nefropatie, cytopenie), reakcje związane z wlewem i zakażenia. Opisano je w punkcie 4.4
Najczęściej występującymi działaniami niepożądanymi związanymi ze stosowaniem produktu leczniczego LEMTRADA (u ≥20% pacjentów) były: wysypka, ból głowy, gorączka i zakażenia dróg oddechowych.
Tabelaryczny wykaz działań niepożądanych
Informacje przedstawione w poniższej tabeli uzyskano na podstawie łącznych danych dotyczących bezpieczeństwa pochodzących od pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg we wszystkich dostępnych okresach obserwacji w ramach badań klinicznych. Działania niepożądane wymieniono według klasyfikacji układów i narządów oraz zgodnie z terminologią MedDRA. Częstość występowania zdefiniowano zgodnie z następującą konwencją: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1000 do <1/100); rzadko (≥1/10 000 do <1/1000 ); bardzo rzadko (<1/10 000); nieznana (częstość nie może być określona na podstawie dostępnych danych). W obrębie każdej grupy o określonej częstości występowania działania niepożądane wymieniono zgodnie ze zmniejszającym się nasileniem.
Tabela 1: Działania niepożądane w badaniu 1., 2., 3. i 4. zaobserwowane u pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg oraz zebrane z obserwacji po dopuszczeniu produktu do obrotu
| Klasyfikacja układów i narządów |
Bardzo często | Często | Niezbyt często | Rzadko | Nieznana |
| Zakażenia i zarażenia pasożytnicze |
Zakażenie górnych dróg oddechowych, zakażenie układu moczowego, zakażenie wirusem opryszczki1 |
Zakażenie wirusem półpaśca2, zakażenia dolnych dróg oddechowych, zapalenie żołądka i jelit, kandydoza jamy ustnej, kandydoza sromu i pochwy, grypa, zapalenie ucha, zapalenie płuc, zakażenie pochwy, zakażenie w obrębie zębów |
Grzybica paznokci, zapalenie dziąseł, grzybicze infekcje skóry, zapalenie migdałków, ostre zapalenie zatok, zapalenie tkanki łącznej, gruźlica, zakażenie wirusem cytomegalii | Listerioza (zapalenie opon mózgowych wywołane przez bakterie z rodzaju Listeria), zakażenie wirusem Epsteina- Barr (EBV) (w tym reaktywacja) | |
| Nowotwory łagodne, złośliwe i nieokreślone (w tym torbiele i polipy) |
Brodawczak skóry |
||||
| Zaburzenia krwi i układu chłonnego | Limfopenia, leukopenia, w tym neutropenia | Limfadenopatia, immunologiczna plamica małopłytkowa, małopłytkowość, niedokrwistość, obniżony hematokryt, leukocytoza | Pancytopenia, niedokrwistość hemolityczna, nabyta hemofilia A | Limfohistiocytoza hemofagocytarna (HLH), zakrzepowa plamica małopłytkowa (TTP) |
|
| Zaburzenia układu immunologi- cznego | Zespół uwolnienia cytokin*, nadwrażliwość, w tym reakcje anafilaktyczne* | Sarkoidoza | |||
| Zaburzenia endokrynolo- giczne |
Choroba Gravesa- Basedowa, nadczynność tarczycy, niedoczynność tarczycy |
Autoimmunologiczne zapalenie tarczycy, w tym podostre zapalenie tarczycy, wole, dodatni wynik badania na obecność przeciwciał przeciwtarczycowych |
|||
| Zaburzenia metabolizmu i odżywiania | Zmniejszone łaknienie | ||||
| Zaburzenia psychiczne | Bezsenność*, lęk, depresja | ||||
| Zaburzenia układu nerwowego |
Ból głowy* | Nawrót stwardnienia rozsianego, zawroty głowy*, niedoczulica, parestezja, drżenie, zaburzenia smaku*, migrena* |
Zaburzenia czucia, przeczulica, napięciowy ból głowy, autoimmunologiczne zapalenie mózgu |
Udar krwotoczny**, rozwarstwienie tętnicy szyjnej i (lub) kręgowej** |
|
| Zaburzenia oka | Zapalenie spojówek, orbitopatia tarczycowa, niewyraźne widzenie |
Podwójne widzenie |
|||
| Zaburzenia ucha i błędnika | Zawroty głowy | Ból ucha | |||
| Zaburzenia serca | Tachykardia* | Bradykardia*, kołatanie serca* |
Migotanie przedsionków* | Niedokrwienie mięśnia sercowego,** zawał mięśnia sercowego** |
|
| Zaburzenia naczyniowe | Zaczerwienie- nie* | Niedociśnienie*, nadciśnienie* | |||
| Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia |
Duszność*, kaszel, krwawienie z nosa, czkawka, ból jamy ustnej i gardła, astma | Ucisk w gardle*, podrażnienie gardła, zapalenie płuc | Krwawienie do światła pęcherzyków płucnych** |
||
| Zaburzenia żołądka i jelit |
Nudności* | Ból brzucha, wymioty, biegunka, niestrawność*, zapalenie jamy ustnej |
Zaparcie, refluks żołądkowo-przełykowy, krwawienie z dziąseł, uczucie suchości w ustach, dysfagia, zaburzenia żołądkowo- jelitowe, hematochezja |
||
| Zaburzenia wątroby i dróg żółciowych | Zwiększona aktywność aminotransferazy asparaginianowej, zwiększona aktywność aminotransferazy alaninowej | Zapalenie pęcherzyka żółciowego, w tym niekamicze zapalenie pęcherzyka żółciowego i ostre niekamicze zapalenie pęcherzyka żółciowego | Autoimmu- nologiczne zapalenie wątroby, zapalenie wątroby (związane z zakażeniem EBV) | ||
| Zaburzenia skóry i tkanki podskórnej |
Pokrzywka*, |
Rumień*, wybroczyny, łysienie, nadmierna potliwość, trądzik, zmiany skórne, zapalenie skóry |
Pęcherze, nocne poty, obrzęk twarzy, egzema, bielactwo nabyte, łysienie plackowate |
||
| Zaburzenia mięśniowo- szkieletowe i tkanki łącznej |
Ból mięśni, osłabienie mięśni, ból stawów, ból pleców, ból kończyn, skurcze mięśni, ból szyi, ból mięśniowo-szkieletowy |
Sztywność mięśniowo- szkieletowa, dyskomfort w obrębie kończyn |
Choroba Stilla u dorosłych |
||
| Zaburzenia nerek i dróg moczowych |
Białkomocz, krwiomocz |
Kamica nerkowa, ketonuria, nefropatie, w tym zespół Goodpasture’a |
|||
| Zaburzenia układu rozrodczego i piersi |
Obfite krwawienia miesiączkowe, nieregularne krwawienia miesiączkowe |
Dysplazja szyjki macicy, brak miesiączki | |||
| Zaburzenia ogólne i stany w miejscu podania | Gorączka*, zmęczenie*, dreszcze* |
Dyskomfort w klatce |
|||
| Badania diagnostyczne | Podwyższony poziom kreatyniny we krwi | Zmniejszenie masy ciała, zwiększenie masy ciała, zmniejszona liczba czerwonych krwinek, dodatni wynik testów bakteryjnych, podwyższony poziom glukozy we krwi, podwyższony wskaźnik MCV | |||
| Urazy, zatrucia i powikłania po zabiegach | Stłuczenia, reakcje związane z wlewem |
1Na zakażenie wirusem opryszczki składają się: opryszczka jamy ustnej (Oral herpes), opryszczka pospolita (Herpes simplex), opryszczka narządów płciowych (Genital herpes), zakażenie wirusem opryszczki (Herpes virus infection), opryszczka pospolita narządów płciowych (Genital herpes simplex), opryszczka skórna (Herpes dermatitis), opryszczka pospolita oczna (Ophthalmic herpes simplex), opryszczka pospolita serologicznie dodatnia (Herpes simplex serology positive)
2Na zakażenie półpaścem składają się: półpasiec (Herpes zoster), półpasiec skórny rozsiany (Herpes zoster cutaneous disseminated), półpasiec oczny (Ophthalmic herpes zoster, Herpes ophthalmic), zakażenie wirusem herpes z zajęciem ośrodkowego układu nerwowego (Herpes zoster infection neurological), zapalenie opon mózgowych (Herpes zoster meningitis).
Opis wybranych działań niepożądanych
Terminy oznaczone w Tabeli 1 gwiazdką (*) obejmują działania niepożądane zgłoszone jako reakcje związane z wlewem.
Terminy oznaczone w Tabeli 1 dwiema gwiazdkami (**) obejmują działania niepożądane obserwowane po dopuszczeniu produktu do obrotu, które w większości przypadków występowały 1-3 dni po podaniu we wlewie produktu leczniczego LEMTRADA, po zastosowaniu którejkolwiek dawki w trakcie cyklu leczenia.
Neutropenia
Zgłaszano przypadki ciężkiej (w tym śmiertelnej) neutropenii w ciągu 2 miesięcy od podania we wlewie produktu leczniczego LEMTRADA.
Profil bezpieczeństwa w długoterminowym okresie obserwacji
Rodzaje działań niepożądanych, z uwzględnieniem stopnia ich nasilenia i ciężkości, zgłaszane u pacjentów leczonych produktem leczniczym LEMTRADA podczas wszystkich dostępnych okresów obserwacji, w tym u pacjentów, którzy otrzymali dodatkowy cykl leczenia, były podobne do tych odnotowanych w ramach badań klinicznych z grupą kontrolną przyjmującą aktywny lek. Reakcje związane z wlewem występowały częściej w 1. cyklu leczenia niż w kolejnych cyklach.
U pacjentów kontynuujących leczenie po zakończeniu badań klinicznych z grupą kontrolną, którzy nie otrzymywali produktu leczniczego LEMTRADA po przyjęciu dwóch początkowych cyklach leczenia, wskaźnik najbardziej niepożądanych działań (liczba zdarzeń niepożądanych/osoborok), które wystąpiły w ciągu 3 do 6 lat był porównywalny lub niższy niż wskaźnik odnotowany w 1. i 2. roku. Wskaźnik działań niepożądanych związanych z tarczycą był najwyższy w 3. roku, a w późniejszym okresie malał.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
W badaniach klinicznych prowadzonych z udziałem grupy kontrolnej dwóm pacjentom chorującym na stwardnienie rozsiane podano przypadkowo w jednym wlewie 60 mg produktu leczniczego LEMTRADA (tj. dawkę całkowitą dla całego początkowego cyklu leczenia), co wywołało u nich poważne reakcje (ból głowy, wysypkę oraz niedociśnienie lub częstoskurcz zatokowy). Dawki produktu leczniczego LEMTRADA większe od przetestowanych w badaniach klinicznych mogą zwiększać natężenie i (lub) wydłużać czas utrzymywania się działań niepożądanych związanych z wlewem lub jej wpływ immunologiczny.
Nie jest znane antidotum w razie przedawkowania alemtuzumabu. Postępowanie obejmuje przerwanie podawania produktu leczniczego i zastosowanie leczenia podtrzymującego.
Grupa farmakoterapeutyczna: Leki immunosupresyjne, Selektywne leki immunosupresyjne, kod ATC: L04AA34.
Mechanizm działania
Alemtuzumab to humanizowane przeciwciało monoklonalne wytwarzane metodą rekombinacji DNA i skierowane przeciw glikoproteinie błony komórkowej CD52 o masie 21–28 kD. Alemtuzumab to przeciwciało IgG1 kappa zawierające regiony zmienne oraz stałe ludzkiego przeciwciała, a także regiony determinujące dopasowanie z mysiego (szczurzego) przeciwciała monoklonalnego. Przybliżona masa cząsteczkowa przeciwciała to 150 kD.
Alemtuzumab wiąże się z antygenem CD52 obecnym w dużych ilościach na powierzchni limfocytów T (CD3+ ) oraz B (CD19+ ), a także, w mniejszych ilościach, na powierzchni komórek NK, monocytów i makrofagów. Ilość antygenu CD52 na powierzchni neutrofilów, komórek plazmatycznych i komórek macierzystych szpiku kostnego jest niewielka lub niewykrywalna. Alemtuzumab działa poprzez cytolizę komórkową zależną od przeciwciał oraz lizę zależną od układu dopełniacza następujące po związaniu z powierzchnią limfocytów T oraz B.
Mechanizm działania terapeutycznego produktu leczniczego LEMTRADA w przypadku stwardnienia rozsianego nie został w pełni wyjaśniony. Jednakże badania wskazują na oddziaływanie o charakterze immunomodulacyjnym w drodze zmniejszenia liczebności i odtworzenia populacji limfocytów, w tym:
- zmianę liczebności, odsetka i właściwości niektórych podgrup limfocytów po leczeniu
- zwiększenie udziału podgrupy limfocytów T regulatorowych
- zwiększenie udziału limfocytów T oraz B pamięci
- przejściowy wpływ na składniki odporności wrodzonej (tj. neutrofile, makrofagi, komórki NK)
Zmniejszenie liczby limfocytów T oraz B w krwioobiegu wywoływane przez produkt leczniczy LEMTRADA i następujące po tym odtworzenie populacji może zmniejszać ryzyko nawrotu choroby, co ostatecznie opóźnia jej progresję.
Działanie farmakodynamiczne
Produkt leczniczy LEMTRADA powoduje zmniejszenie liczby limfocytów T oraz B w krwioobiegu po każdym cyklu leczenia, przy czym najniższa liczebność tych komórek występuje 1 miesiąc po zakończeniu cyklu leczenia (najwcześniejszy punkt czasowy po zakończeniu leczenia w badaniach fazy III). Liczebność limfocytów wzrasta ponownie w miarę upływu czasu, a odtworzenie populacji limfocytów B zostaje zwykle osiągnięte w ciągu 6 miesięcy. Liczba limfocytów CD3+ i CD4+ rośnie znacznie wolniej, jednak zazwyczaj nie powraca do wartości początkowych przed upływem 12 miesięcy od zakończenia leczenia. U około 40% pacjentów całkowita liczba limfocytów osiągała dolną granicę normy (DGN) w ciągu 6 miesięcy po każdym cyklu leczenia, a u około 80% pacjentów całkowita liczba limfocytów osiągała DGN w ciągu 12 miesięcy po każdym cyklu leczenia.
Wpływ produktu leczniczego LEMTRADA na neutrofile, monocyty, eozynofile, bazofile i komórki NK ma jedynie charakter przejściowy.
Skuteczność kliniczna i bezpieczeństwo stosowania
Bezpieczeństwo stosowania i skuteczność alemtuzumabu u pacjentów ze stwardnieniem rozsianym oceniano w 3 randomizowanych badaniach klinicznych z użyciem czynnego produktu porównawczego prowadzonych metodą zaślepienia badacza oceniającego oraz w 1 przedłużonym badaniu klinicznym prowadzonym metodą zaślepienia oceniającego wyniki badacza oraz bez grupy kontrolnej z udziałem pacjentów chorujących na RRMS.
Dla badania 1., 2., 3., i 4. założenia/dane demograficzne przedstawiono w Tabeli 2.
|
Tabela 2: Projekt badania i charakterystyka w punkcie wyjściowym badania 1. 2., 3. i 4. |
|||
|
|
Badanie 1. |
Badanie 2. |
Badanie 3. |
|
Nazwa badania |
CAMMS323(CARE-MS I) |
CAMMS32400507(CARE-MS II) |
CAMMS223 |
|
Założenia badania |
Z grupą kontrolną, randomizowane, prowadzone metodą zaślepienia oceniającego wyniki badacza |
Z grupą kontrolną, randomizowane, prowadzone metodą zaślepienia oceniającego wyniki badacza i zaślepienia dawki |
Z grupą kontrolną, randomizowane, prowadzone metodą zaślepienia oceniającego wyniki badacza |
|
Wywiad chorobowy |
Pacjenci z aktywnym stwardnieniem rozsianym definiowanym jako wystąpienieco najmniej 2 nawrotów choroby w ciągu ostatnich 2 lat. |
Pacjenci z aktywnym stwardnieniem rozsianym definiowanym jako wystąpienie conajmniej 2 nawrotów choroby w ciągu ostatnich 2 lat oraz z 1 lub więcej ogniskami wzmocnienia po podaniu kontrastu |
|
|
Czas trwania |
2 lata |
3 lata |
|
|
Populacja badania |
Pacjenci wcześniej nieleczeni |
Pacjenci z niewystarczającą odpowiedzią nawcześniejsze leczenie* |
Pacjenci wcześniej nieleczeni |
|
Charakterystyka w punkcie wyjściowym badania |
|
|
|
|
Średni wiek (lata) |
33 |
35 |
32 |
|
Średni czas/mediana czasu trwania choroby |
2 /1,6 lat |
4,5/3,8 lat |
1,5/1,3 lat |
|
Średni czas trwania wcześniejszego leczenia stwardnienia rozsianego (stosowany≥1 lek) |
Brak leczenia |
36 miesięcy |
Brak leczenia |
|
Odsetek pacjentów, którzy odbyli ≥2 wcześniejsze leczenia stwardnienia rozsianego |
Nie dotyczy |
28% |
Nie dotyczy |
|
Średni wynik w skali EDSS w punkciepoczątkowym badania |
2,0 |
2,7 |
1,9 |
|
|
Badanie 4. |
||
|
Nazwa badania |
CAMMS03409 |
||
|
Założenia badania |
Przedłużone badanie kliniczne prowadzone metodą zaślepienia badacza i bez grupy kontrolnej |
||
|
Badana populacja |
Pacjenci, którzy uczestniczyli w badaniach: CAMMS223, CAMMS323lub CAMMS32400507 |
||
|
Czas trwania przedłużonego badania |
4 lata |
||
* Określani jako pacjenci, u których w trakcie leczenia interferonem beta lub octanem glatirameru, po co najmniej 6 miesiącach terapii, doszło do co najmniej 1 nawrotu choroby.
‡ Pierwszorzędowy punkt końcowy badania został oceniony po 3 latach. Mediana dodatkowego okresu obserwacji, podczas którego gromadzono dane, wynosiła 4,8 roku (maksymalnie 6,7 roku).
Wyniki badania 1. i 2. przedstawiono w Tabeli 3.
|
Tabela 3: Kluczowe punkty kliniczne i końcowe stwierdzane w ramach NMR w badaniach 1. i 2. |
||||
|
|
Badanie 1. |
Badanie 2. |
||
|
Nazwa badania |
CAMMS323 |
CAMMS32400507 |
||
|
Kliniczne punkty końcowe |
LEMTRADA |
Podawany podskórnie IFNB-1a |
LEMTRADA |
Podawany podskórnie IFNB-1a |
|
Częstość występowania nawrotów choroby1 Roczny wskaźnik nawrotów (ARR) (95% CI) |
0,18 |
0,39 |
0,26 |
0,52 |
|
Współczynnik częstości (95% CI) Redukcja ryzyka |
0,45 (0,32; 0,63) |
0,51 (0,39; 0,65) |
||
|
Niepełnosprawność1(potwierdzona progresja niepełnosprawności[CDW])2 Liczba pacjentów z CDW trwającym 6 miesięcy (%) (95% CI) |
8,0% |
11,1% |
12,7% |
21,1% |
|
Współczynnik ryzyka (95% CI) |
0,70 (0,40; 1,23) |
0,58 (0,38; 0,87) |
||
|
Pacjenci, u których w roku 2. nie doszło do nawrotu choroby |
77,6% |
58,7% |
65,4% |
46,7% |
|
Zmiana wyniku w skali EDSS w roku 2. w porównaniu z punktem początkowym badania3 |
-0,14(-0,25; -0,02) |
-0,14 (-0,29; 0,01) |
-0,17 (-0,29; -0,05) |
0,24 (0,07; 0,41) |
|
Punkty końcowe stwierdzane w badaniu NMR (0–2 lat) |
||||
|
Mediana % zmiany objętości zmiany chorobowej w sekwencji T2 badania NMR |
-9,3 (-19,6; -0,2) |
-6,5 (-20,7; 2,5) |
-1,3 |
-1,2 |
|
Pacjenci z nowymi lub powiększającymi się zmianami chorobowymi w sekwencji T2 w roku 2. |
48,5% |
57,6% |
46,2% |
67,9% |
|
Pacjenci ze zmianami chorobowymi pochłaniającymi gadolin w roku 2. |
15,4% |
27,0% |
18,5% |
34,2% |
|
Pacjenci z nowymi hipointensywnymi zmianami chorobowymi w sekwencji T1 wroku 2. |
24,0% |
31,4% |
19,9% |
38,0% |
|
Mediana % zmiany w miąższowej frakcji mózgu |
-0,867 |
-1,488 |
-0,615 |
-0,810 |
Rycina 1: Czas do wystąpienia potwierdzonej progresji niepełnosprawności utrzymującej się przez 6 miesięcy w badaniu 2.
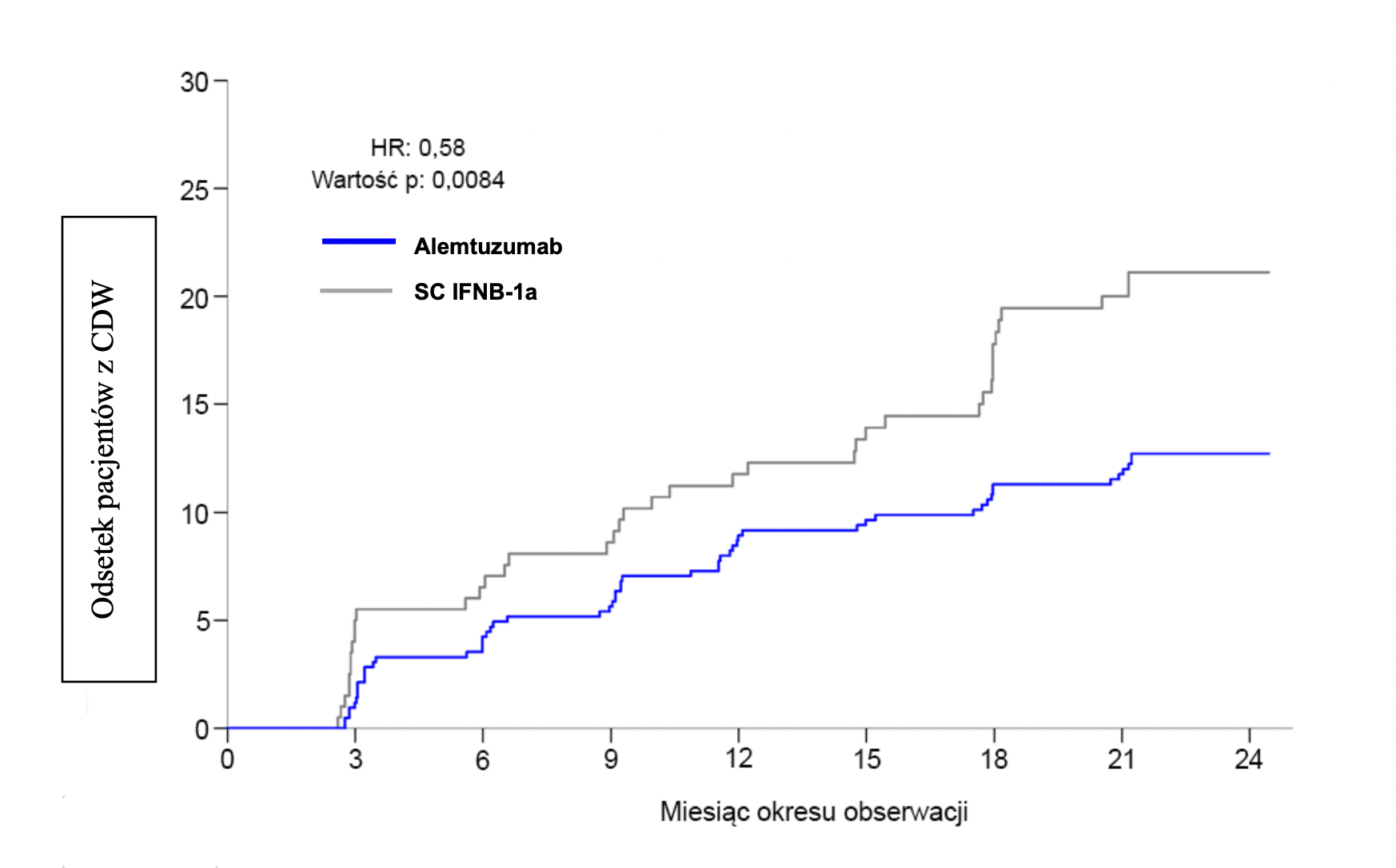
Nasilenie nawrotów choroby
Analizy dodatkowe danych z badania 1. (CAMMS323) łącznie z wpływem na częstość występowania nawrotów choroby wykazały, że w porównaniu ze stosowaniem IFNB-1a, po podaniu produktu leczniczego LEMTRADA w dawce 12 mg na dobę u znacznie mniejszej liczby pacjentów (obniżenie o 61%, p = 0,0056) wystąpiły ciężkie nawroty choroby oraz znacząco mniejsza liczba nawrotów wymagała leczenia steroidami (obniżenie o 58%, p <0,0001). Analizy dodatkowe danych z badania 2. (CAMMS32400507) wykazały, że w porównaniu ze stosowaniem IFNB-1a po podaniu produktu leczniczego LEMTRADA w dawce 12 mg na dobę u znacznie mniejszej liczby pacjentów (obniżenie o 48%, p = 0,0121) wystąpiły ciężkie nawroty choroby oraz znacznie mniej nawrotów wymagało leczenia steroidami (obniżenie o 56%, p <0,0001) lub hospitalizacji (obniżenie o 55%, p = 0,0045).
Potwierdzone zmniejszenie niepełnosprawności (CDI)
Czas do wystąpienia CDI (potwierdzonego zmniejszenia niepełnosprawności) definiowano jako spadek o co najmniej jeden punkt na rozszerzonej skali oceny stanu niepełnosprawności (EDSS) od wartości bazowej ≥2 utrzymujący się przez co najmniej 6 miesięcy. Wartość CDI to miara utrzymującego się zmniejszenia niepełnosprawności. U 29% pacjentów leczonych produktem leczniczym LEMTRADA w badaniu 2. uzyskano CDI, natomiast w przypadku pacjentów, którym podawano podskórnie IFNB-1a, ten punkt końcowy osiągnęło zaledwie 13% pacjentów. Różnica była statystycznie istotna (p= 0,0002).
W badaniu 3. (badanie CAMMS223 fazy II) przez 3 lata oceniano bezpieczeństwo stosowania i skuteczność produktu leczniczego LEMTRADA u pacjentów z RRMS. W momencie włączenia do badania pacjenci uzyskali wynik na skali EDSS mieszczący się w przedziale 0–3,0, stwierdzono u nich co najmniej dwa epizody kliniczne stwardnienia rozsianego w ostatnich 2 latach i ≥1 zmianę chorobową pochłaniającą gadolin. Pacjenci nie byli wcześniej leczeni z powodu stwardnienia rozsianego. Zastosowano u nich leczenie produktem leczniczym LEMTRADA w dawce 12 mg na dobę (N = 108) lub 24 mg na dobę (N = 108), który podawano raz dziennie przez 5 dni w miesiącu 0. oraz przez 3 dni w miesiącu 12., lub podawanym podskórnie IFNB-1a w dawce 44 µg (N = 107) 3 razy w tygodniu przez 3 lata. U czterdziestu sześciu pacjentów przeprowadzono trzeci cykl leczenia produktem leczniczym LEMTRADA, który podawano w dawce 12 mg na dobę lub 24 mg na dobę przez 3 dni w miesiącu 24.
Po upływie 3 lat produkt leczniczy LEMTRADA spowodował obniżenie ryzyka wystąpienia potwierdzonej progresji niepełnosprawności trwającej 6 miesięcy o 76% (współczynnik ryzyka 0,24 [95% CI: 0,110; 0,545], p <0,0006) i rocznego wskaźnika nawrotów (ARR) o 67% (współczynnik częstości 0,33 [95% CI: 0,196; 0,552], p <0,0001) w porównaniu z podawanym podskórnie IFNB-1a. Produkt leczniczy LEMTRADA podawany w dawce 12 mg na dobę prowadził do znacząco niższych wyników na skali EDSS (poprawy w porównaniu z wynikami w punkcie początkowym badania) w czasie 2 lat obserwacji w porównaniu z IFNB-1a (p <0,0001).
W podgrupie pacjentów z RRMS z 2 lub więcej rzutami w poprzednim roku i co najmniej 1 zmianą T1 ulegającą wzmocnieniu po podaniu gadolinu w punkcie początkowym, roczny wskaźnik rzutów wynosił 0,26 (95% CI: 0,20; 0,34) w grupie otrzymującej produkt leczniczy Lemtrada (n = 205) i 0,51 (95% CI: 0,40; 0,64) w grupie IFNB-1a (n = 102) (p <0,0001). Analiza obejmuje wyłącznie dane z badań fazy III (CAMMS324 i CAMMS323) ze względu na różnice w algorytmach rezonansu magnetycznego między badaniami fazy II i fazy III. Wyniki uzyskano z analizy post hoc i należy je interpretować z ostrożnością.
Dane na temat długotrwałej skuteczności
Badanie 4. było wieloośrodkowym, prowadzonym metodą otwartej próby z zaślepieniem oceniającego wyniki badacza, przedłużonym badaniem skuteczności i bezpieczeństwa fazy III mającym na celu ocenę długotrwałej skuteczności i bezpieczeństwa stosowania produktu leczniczego LEMTRADA u pacjentów z RRMS, którzy uczestniczyli wcześniej w badaniu 1., 2., lub 3. (wcześniejsze badania fazy III i II). Badanie to dostarczyło danych na temat skuteczności i bezpieczeństwa w okresie, którego mediana wynosiła 6 lat od momentu rozpoczęcia udziału w badaniu 1 i 2. Pacjenci biorący udział w badaniu przedłużonym (badanie 4.) zostali zakwalifikowani do dodatkowego(-wych) cyklu(-ów) leczenia produktem leczniczym LEMTRADA doraźnie na podstawie udokumentowanej wznowionej aktywności choroby, zdefiniowanej jako wystąpienie co najmniej jednego rzutu stwardnienia rozsianego i (lub) pojawienie się co najmniej dwóch nowych lub powiększających się zmian w mózgowiu lub kręgosłupie widocznych w badaniu rezonansem magnetycznym (MRI). Dodatkowy(-e) cykl(e) leczenia produktem leczniczym LEMTRADA obejmował(y) podanie leku w dawce 12 mg na dobę przez 3 kolejne dni (dawka całkowita 36 mg) co najmniej 12 miesięcy po wcześniejszym cyklu leczenia.
91,8% pacjentów leczonych produktem leczniczym LEMTRADA w dawce 12 mg w ramach badania 1. i 2. wzięło udział w badaniu 4., a 82,7% z nich ukończyło badanie. Około połowa (51,2%) pacjentów przyjmujących wstępnie produkt leczniczy LEMTRADA w dawce 12 mg na dobę w badaniu 1. i 2. i którzy uczestniczyli w badaniu 4., otrzymała tylko 2 początkowe cykle leczenia produktem LEMTRADA i nie otrzymała żadnego innego rodzaju leczenia modyfikującego przebieg choroby przez 6 lat okresu obserwacji.
46,6% pacjentów początkowo leczonych produktem leczniczym LEMTRADA w dawce 12 mg na dobę w ramach badania 1. i 2. otrzymało dodatkowy cykl leczenia na podstawie udokumentowanej oznaki wznowionej aktywacji choroby (kolejny rzut lub wynik badania MRI) oraz decyzji podjętej przez lekarza prowadzącego. W momencie rozpoczęcia badania żadna cecha nie była wyznacznikiem tego, którzy pacjenci otrzymają później jeden lub więcej dodatkowych cyklów leczenia.
Przez 6 lat od rozpoczęcia leczenia produktem leczniczym LEMTRADA u pacjentów, u których kontynuowano obserwację, wykazano, że współczynniki nawrotu stwardnienia rozsianego, powstawania uszkodzeń mózgu widocznych w badaniu rezonansem magnetycznym (MRI) i utraty objętości tkanki mózgowej są zgodne z efektami leczenia produktem leczniczym LEMTRADA w ramach badania 1. i 2., podobnie jak przeważnie stabilne lub poprawione wyniki oceny niepełnosprawności. Uwzględniając dalszą obserwację w badaniu 4, u pacjentów przyjmujących produkt leczniczy LEMTRADA w badaniu 1. i 2. roczny wskaźnik nawrotów (ARR) wynosił, odpowiednio, 0,17 i 0,23, potwierdzona progresja niepełnosprawności (CDW) 22,3% i 29,7%, a u 32,7% i 42,5% pacjentów uzyskano potwierdzone zmniejszenie niepełnosprawności (CDI). W każdym roku badania 4. pacjenci biorący udział w obydwu badaniach stale wykazywali niskie ryzyko powstawania nowych ognisk podwyższonego sygnału w obrazach T2-zależnych (27,4-33,2%) lub zmian chorobowych pochłaniających gadolin (9,4-13,5%), a mediana rocznej procentowej zmiany frakcji miąższu mózgu wynosiła od 0,19% do -0,09%.
U pacjentów, którzy otrzymali jeden lub dwa dodatkowe cykle leczenia produktem leczniczym LEMTRADA, zaobserwowano poprawę współczynnika nawrotu choroby, aktywności mózgu w badaniu MRI i średnich wyników niepełnosprawności w wyniku pierwszego lub drugiego ponownego podania produktu leczniczego LEMTRADA (cykle leczenia 3. i 4.), w porównaniu do wyników z roku poprzedzającego. U tych pacjentów roczny wskaźnik nawrotów (ARR) zmalał z 0,79 w roku poprzedzającym 3. cykl leczenia do 0,18 w kolejnym roku. Średni wynik w skali EDSS zmniejszył się z 2,89 do 2,69. Odsetek pacjentów z nowymi lub powiększającymi się zmianami w obrazach T2-zależnych zmniejszył się z 50,8% w roku poprzedzającym 3. cykl leczenia do 35,9% rok później. Odsetek pacjentów z nowymi lub powiększającymi się zmianami pochłaniającymi gadolin zmniejszył się z 32,2% do 11,9%. Podobną poprawę rocznego wskaźnika nawrotów (ARR), średniego wyniku w skali EDSS oraz zmian w obrazach T2-zależnych i zmian pochłaniających gadolin odnotowano po 4. cyklu leczenia w porównaniu z rokiem go poprzedzającym. Te wyniki utrzymywały się w późniejszym czasie, ale nie można wyciągnąć żadnych zdecydowanych wniosków dotyczących długotrwałego stosowania (np. 3 lub 4 lata po dodatkowych cyklach leczenia), ponieważ wielu pacjentów zakończyło badanie przed osiągnięciem tych punktów czasowych.
Nie określono w pełni korzyści i zagrożeń związanych ze stosowaniem 5 lub więcej cyklów leczenia.
Immunogenność
Podobnie jak w przypadku wszystkich białek terapeutycznych, może wystąpić immunogenność. Dane odzwierciedlają odsetek pacjentów, u których stwierdzono dodatni wynik badania na obecność przeciwciał przeciw alemtuzumabowi wykonanego metodą testu immunoabsorpcji enzymatycznej (ELISA) i potwierdzonego testem wiązania kompetycyjnego. Próbki z wynikami dodatnimi oceniano dalej z wykorzystaniem cytometrii przepływowej pod kątem obecności dowodów na hamowanie aktywności in vitro. Od pacjentów ze stwardnieniem rozsianym biorących udział w badaniach klinicznych pobrano próbki surowicy w 1., 3. i 12. miesiącu po każdym cyklu leczenia w celu wykrycia przeciwciał przeciw alemtuzumabowi. U około 85% pacjentów otrzymujących produkt leczniczy LEMTRADA stwierdzono w trakcie badania dodatni wynik testu na obecność przeciwciał przeciw alemtuzumabowi, a u ≥90% z nich uzyskano również dodatni wynik na obecność przeciwciał hamujących wiązanie alemtuzumabu in vitro. U pacjentów, u których doszło do wytworzenia przeciwciał przeciw alemtuzumabowi, nastąpiło to w ciągu 15 miesięcy po początkowej ekspozycji. Podczas dwóch cyklów leczenia nie stwierdzono związku między obecnością przeciwciał przeciw alemtuzumabowi lub przeciwciał przeciw alemtuzumabowi hamujących jego wiązanie a zmniejszeniem skuteczności działania produktu leczniczego, zmianą właściwości farmakodynamicznych lub wystąpieniem działań niepożądanych, w tym reakcji związanych z wlewem. Wysokie miano przeciwciał przeciw alemtuzumabowi zaobserwowane u niektórych pacjentów miało związek z niecałkowitym zmniejszeniem liczby limfocytów po 3. lub 4. cyklu leczenia, ale nie wykazano wyraźnego wpływu przeciwciał przeciw alemtuzumabowi na kliniczną skuteczność i profil bezpieczeństwa produktu leczniczego LEMTRADA. Występowanie przeciwciał jest bardzo uzależnione od czułości i swoistości badania. Dodatkowo na obserwowane w badaniu występowanie dodatnich wyników na obecność przeciwciał (w tym przeciwciał hamujących) może wpływać kilka czynników, w tym metodologia badania, obchodzenie się z próbką, moment pobrania próbki, jednocześnie stosowane produkty lecznicze i choroby współwystępujące. Z tego powodu porównywanie występowania przeciwciał przeciw produktowi leczniczemu LEMTRADA z występowaniem przeciwciał przeciw innym produktom może być mylące.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań alemtuzumabu u dzieci w wieku od urodzenia do 10 roku życia w leczeniu stwardnienia rozsianego (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego LEMTRADA w jednej lub kilku podgrupach populacji dzieci i młodzieży leczonych z powodu RRMS (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Właściwości farmakokinetyczne alemtuzumabu oceniono w grupie 216 pacjentów z RRMS, którzy otrzymywali dożylne wlewy zawierające produkt leczniczy w dawce 12 mg na dobę lub 24 mg na dobę przez 5 kolejnych dni, a następnie przez 3 kolejne dni po 12 miesiącach od początkowego cyklu leczenia. Stężenia w surowicy rosły wraz z każdą kolejną dawką w ramach cyklu leczenia, a najwyższe stężenia obserwowano po ostatnim wlewie w cyklu. Podanie dawki 12 mg na dobę skutkowało uzyskaniem średniej wartości Cmax wynoszącej 3014 ng/ml w 5. dniu początkowego cyklu leczenia oraz 2276 ng/ml w 3. dniu drugiego cyklu leczenia. Okres połowicznego rozpadu alfa oszacowano na 4–5 dni. Wartość ta była porównywalna pomiędzy cyklami leczenia i skutkowała niskimi lub niewykrywalnymi stężeniami w surowicy po około 30 dniach od zakończenia każdego cyklu leczenia.
Alemtuzumab to białko, którego oczekiwana ścieżka przemian metabolicznych obejmuje degradację do małych peptydów i poszczególnych aminokwasów w drodze działania szeroko rozpowszechnionych enzymów proteolitycznych. Nie przeprowadzono klasycznych badań dotyczących biotransformacji.
Na postawie dostępnych danych nie można wyciągnąć wniosków na temat wpływu rasy ani płci na właściwości farmakokinetyczne alemtuzumabu. Nie badano właściwości farmakokinetycznych alemtuzumabu u pacjentów w wieku 55 lat ani starszych chorujących na RRMS.
Rakotwórczość i mutageneza
Nie przeprowadzono badań w celu oceny potencjału rakotwórczego i mutagennego alemtuzumabu.
Płodność i reprodukcja
Leczenie alemtuzumabem podawanym dożylnie w dawkach do 10 mg/kg na dobę przez 5 kolejnych dni (wartość AUC o 7,1 razy wyższa niż ekspozycja u ludzi przy zalecanej dawce dobowej) nie miało wpływu na płodność i reprodukcję u samców transgenicznych myszy huCD52. Liczba prawidłowo zbudowanych plemników była znacznie mniejsza (<10%) niż w grupie kontrolnej, a odsetek nieprawidłowych plemników (z odłączoną główką lub bez główki) uległ znacznemu zwiększeniu (do 3%). Jednak te zmiany nie miały wpływu na płodność i z tego powodu nie zostały uznane za niekorzystne.
U samic myszy, którym podawano alemtuzumab dożylnie w dawkach do 10 mg/kg na dobę (wartość AUC o 4,7 razy wyższa niż ekspozycja u ludzi przy zalecanej dawce dobowej) przez 5 kolejnych dni przed dopuszczeniem samca typu dzikiego, średnia liczba ciałek żółtych i miejsc implantacji na mysz uległa znacznemu zmniejszeniu w porównaniu ze zwierzętami, którym podawano jedynie nośnik. U ciężarnych myszy, którym podawano dawkę 10 mg/kg na dobę, obserwowano zmniejszony przyrost masy ciążowej w porównaniu z grupą kontrolną, w której podawano jedynie nośnik.
Badanie toksyczności reprodukcyjnej u ciężarnych myszy, którym podawano alemtuzumab dożylnie w dawkach do 10 mg/kg na dobę (wartość AUC o 2,4 razy wyższa niż ekspozycja u ludzi przy zalecanej dawce dobowej wynoszącej 12 mg na dobę) przez 5 kolejnych dni ciąży, ujawniło znaczny wzrost liczby samic, u których doszło do obumarcia lub resorpcji wszystkich zarodków, z jednoczesnym zmniejszeniem liczby samic z żywymi płodami. Po dawkach do 10 mg/kg na dobę nie obserwowano żadnych nieprawidłowości ani zmian budowy zewnętrznej, a także w obrębie tkanek miękkich czy szkieletu.
U myszy w czasie ciąży i po porodzie obserwowano przenikanie przez łożysko i potencjalną aktywność farmakologiczną alemtuzumabu. W badaniach prowadzonych na myszach u młodych wystawionych w okresie płodowym na działanie alemtuzumabu w dawce 3 mg/kg na dobę przez 5 kolejnych dni (wartość AUC 0,6 razy wyższa niż ekspozycja u ludzi przy zalecanej dawce dobowej wynoszącej 12 mg na dobę) obserwowano zmiany liczby limfocytów. Rozwój poznawczy, fizyczny i płciowy młodych wystawionych na działanie alemtuzumabu w czasie laktacji nie był zakłócany przy dawkach do 10 mg/kg na dobę.
Disodu fosforan dwuwodny (E339)
Disodu edetynian dwuwodny
Potasu chlorek (E508)
Potasu diwodorofosforan (E340)
Polisorbat 80 (E433)
Sodu chlorek
Woda do wstrzykiwań
Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. Nie dotyczy to produktów leczniczych wymienionych w punkcie 6.6.
Koncentrat
4 lata
Roztwór rozcieńczony
Stabilność chemiczna i fizyczna produktu leczniczego jest zachowywana przez 8 godzin w temperaturze od 2°C do 8°C.
Z mikrobiologicznego punktu widzenia zaleca się natychmiastowe wykorzystanie produktu leczniczego. Jeśli nie zostanie on wykorzystany od razu, za warunki i czas przechowywania do momentu jego zastosowania odpowiada osoba podająca lek. Nie należy jednak przechowywać roztworu dłużej niż 8 godzin w temperaturze od 2°C do 8°C, w miejscu chronionym przed światłem.
Koncentrat
Przechowywać w lodówce (2°C–8°C).
Nie zamrażać.
Przechowywać fiolkę w opakowaniu zewnętrznym w celu ochrony przed światłem.
Warunki przechowywania produktu leczniczego po rozcieńczeniu, patrz punkt 6.3.
Produkt leczniczy LEMTRADA jest dostarczany w przezroczystej szklanej fiolce o pojemności 2 ml z korkiem z gumy butylowej i aluminiowym wieczkiem zabezpieczającym ze zrywalną częścią plastikową.
Wielkość opakowań: pudełko tekturowe z 1 fiolką.
Zawartość fiolki należy przed podaniem sprawdzić pod kątem obecności cząstek i zmiany zabarwienia. Nie należy stosować produktu leczniczego w przypadku stwierdzenia obecności cząstek lub zmiany barwy koncentratu.
Fiolek nie należy wstrząsać przed użyciem.
W przypadku podania dożylnego należy pobrać z fiolki do strzykawki z zachowaniem zasad aseptyki 1,2 ml produktu leczniczego LEMTRADA. Następnie należy dodać go do 100 ml roztworu chlorku sodu o stężeniu 9 mg/ml (0,9%) lub roztworu glukozy (5%) do infuzji. Tego produktu leczniczego nie wolno rozcieńczać innymi rozpuszczalnikami. Worek należy delikatnie odwrócić w celu wymieszania roztworu.
Należy zwrócić szczególną uwagę na zapewnienie jałowych warunków przygotowywania roztworu. Zaleca się natychmiastowe podanie rozcieńczonego produktu. Każda fiolka jest przeznaczona wyłącznie do jednorazowego użytku.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Sanofi Belgium
Leonardo Da Vincilaan 19
B-1831 Diegem
Belgia
EU/1/13/869/001
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 12 września 2013
Data wydania przedłużenia pozwolenia: 02 lipca 2018
Szczegółowe informacje o tym produkcie leczniczym są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu.
Powered by Biogen Poland Sp. z o.o.
(Biogen-101710)
 i
i  "Dodaj aplikację do ekranu początkowego"
"Dodaj aplikację do ekranu początkowego"