Powered by Biogen Poland Sp. z o.o.
Powered by Biogen Poland Sp. z o.o.
Kesimpta 20 mg roztwór do wstrzykiwań w ampułko-strzykawce
Kesimpta 20 mg roztwór do wstrzykiwań we wstrzykiwaczu
Kesimpta 20 mg roztwór do wstrzykiwań w ampułko-strzykawce
Każda ampułko-strzykawka zawiera 20 mg ofatumumabu w 0,4 ml roztworu (50 mg/ml).
Kesimpta 20 mg roztwór do wstrzykiwań we wstrzykiwaczu
Każdy wstrzykiwacz zawiera 20 mg ofatumumabu w 0,4 ml roztworu (50 mg/ml).
Ofatumumab jest w pełni ludzkim przeciwciałem monoklonalnym wytwarzanym w mysiej linii komórkowej (NS0) metodą rekombinacji DNA.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Roztwór do wstrzykiwań
Roztwór do wstrzykiwań we wstrzykiwaczu (wstrzykiwacz Sensoready)
Roztwór jest przezroczysty do lekko opalizującego oraz bezbarwny do lekko brązowawożółtego.
Produkt leczniczy Kesimpta jest wskazany do stosowania w leczeniu dorosłych pacjentów z rzutowymi postaciami stwardnienia rozsianego (ang. relapsing forms of multiple sclerosis, RMS) z aktywną chorobą potwierdzoną w badaniu klinicznym lub obrazowym (patrz punkt 5.1).
Leczenie powinno być rozpoczynane przez lekarza mającego doświadczenie w leczeniu chorób neurologicznych.
Dawkowanie
Zalecana dawka to 20 mg ofatumumabu podawana we wstrzyknięciu podskórnym:
Pominięcie dawki
W razie pominięcia wstrzyknięcia należy podać je tak szybko, jak to możliwe, nie czekając do terminu kolejnej zaplanowanej dawki. Następne dawki należy podać, zachowując zalecane odstępy.
Specjalne populacje pacjentów
Dorośli w wieku powyżej 55 lat
Nie przeprowadzono badań z udziałem pacjentów z SM w wieku powyżej 55 lat. Na podstawie dostępnych ograniczonych danych można uznać, że u pacjentów w wieku powyżej 55 lat dostosowanie dawkowania nie jest konieczne (patrz punkt 5.2).
Zaburzenia czynności nerek
U pacjentów z zaburzeniami czynności nerek modyfikacja dawki nie wydaje się konieczna (patrz punkt 5.2).
Zaburzenia czynności wątroby
U pacjentów z zaburzeniami czynności wątroby modyfikacja dawki nie wydaje się konieczna (patrz punkt 5.2).
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności produktu leczniczego Kesimpta u dzieci w wieku od 0 do 18 lat. Dane nie są dostępne.
Sposób podawania
Ten produkt leczniczy jest przeznaczony do samodzielnego podawania przez pacjenta we wstrzyknięciu podskórnym.
Do typowych miejsc wstrzyknięć podskórnych należą brzuch, udo i zewnętrzna górna część ramienia.
Pierwsze wstrzyknięcie powinno zostać podane pod nadzorem osoby należącej do fachowego personelu medycznego (patrz punkt 4.4).
Szczegółową instrukcję podawania produktu zamieszczono w ulotce dla pacjenta.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Pacjenci w stanie silnie obniżonej odporności (patrz punkt 4.4).
Ciężka aktywna infekcja do czasu jej ustąpienia (patrz punkt 4.4).
Znany aktywny nowotwór złośliwy.
Identyfikowalność
W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Reakcje związane ze wstrzyknięciem
Należy poinformować pacjentów, że mogą wystąpić reakcje związane ze wstrzyknięciem (ogólnoustrojowe, ang. systemic injection-related reactions, SIRRs), przeważnie w ciągu 24 godzin i głównie po pierwszym wstrzyknięciu (patrz punkt 4.8). W badaniach klinicznych nad RMS stwierdzano jedynie ograniczone korzyści ze stosowania premedykacji steroidami. Jeśli wystąpią reakcje związane ze wstrzyknięciem, można stosować leczenie objawowe. W związku z tym stosowanie premedykacji nie jest konieczne.
Objawy reakcji w miejscu wstrzyknięcia (miejscowe) obserwowane w badaniach klinicznych obejmowały rumień, obrzęk, swędzenie i ból (patrz punkt 4.8). Objawy najczęściej obserwowane w badaniach nad RMS obejmują gorączkę, ból głowy, ból mięśni, dreszcze, uczucie zmęczenia, nudności i wymioty, a ich nasilenie było przeważnie (99,8%) łagodne do umiarkowanego. W badaniach klinicznych nad RMS nie zgłaszano SIRRs zagrażających życiu (patrz punkt 4.8).
Niektóre objawy SIRR mogą być klinicznie nie do odróżnienia od ostrych reakcji nadwrażliwości typu 1 (IgE-zależnych). Reakcja nadwrażliwości może wystąpić podczas każdego wstrzyknięcia, chociaż zazwyczaj nie zdarza się po pierwszym wstrzyknięciu. W przypadku kolejnych wstrzyknięć, objawy poważniejsze niż występujące wcześniej lub nowe ciężkie objawy powinny skłonić do rozważenia potencjalnej reakcji nadwrażliwości. Nie wolno leczyć ofatumumabem pacjentów ze znaną nadwrażliwością IgE-zależną na ofatumumab. (patrz punkt 4.3).
Dodatkowe SIRRs zgłaszane w okresie po wprowadzeniu do obrotu obejmowały wysypkę, pokrzywkę, duszność i obrzęk naczynioruchowy (np. opuchnięcie języka, gardła lub krtani) i, w rzadkich przypadkach, anafilaksję. Chociaż wystąpiły przypadki poważne, skutkujące przerwaniem leczenia ofatumumabem, odnotowano także przypadki poważne, w których pacjenci byli w stanie kontynuować leczenie ofatumumabem bez dalszych incydentów SIRRs.
Pierwsze wstrzyknięcie powinno zostać podane pod nadzorem odpowiednio przeszkolonej osoby należącej do fachowego personelu medycznego (patrz punkt 4.2).
Zakażenia
Zaleca się ocenę stanu odporności pacjenta przed rozpoczęciem leczenia.
Na podstawie sposobu działania i dostępnego doświadczenia klinicznego ofatumumab może zwiększać ryzyko wystąpienia zakażeń (patrz punkt 4.8).
Należy odroczyć podanie produktu u pacjentów z aktywnym zakażeniem do czasu jego ustąpienia.
Nie wolno podawać ofatumumabu pacjentom w stanie silnie obniżonej odporności (np. ze znaczną neutropenią lub limfopenią).
Postępująca wieloogniskowa leukoencefalopatia
Ponieważ u pacjentów leczonych przeciwciałami anty-CD20, innymi lekami stosowanymi w SM i ofatumumabem podawanym w znacznie większych dawkach we wskazaniach onkologicznych obserwowano zakażenia wirusem Johna Cunninghama (JC) powodujące postępującą wieloogniskową leukoencefalopatię (ang. progressive multifocal leukoencephalopathy, PML), lekarze powinni zwracać uwagę na występowanie PML w wywiadzie oraz wszelkie objawy kliniczne lub wyniki badań obrazowych MRI (obrazowanie rezonansem magnetycznym), które mogą sugerować PML. W razie podejrzenia PML, leczenie ofatumumabem należy wstrzymać do czasu wykluczenia PML.
Reaktywacja wirusowego zapalenia wątroby typu B
U pacjentów leczonych przeciwciałami anty-CD20 występowała reaktywacja wirusowego zapalenia wątroby typu B, co w niektórych przypadkach powodowało piorunujące zapalenie wątroby, niewydolność wątroby i zgon.
Pacjentów z aktywnym zapaleniem wątroby typu B nie należy leczyć ofatumumabem. Przed rozpoczęciem leczenia u wszystkich pacjentów należy wykonać badania przesiewowe w kierunku WZW B. W ramach niezbędnego minimum, badania przesiewowe powinny obejmować sprawdzenie obecności antygenu powierzchniowego wirusa zapalenia wątroby typu B (HBsAg) i przeciwciał przeciwko antygenowi rdzeniowemu wirusa zapalenia wątroby typu B (HBcAb). Można je uzupełnić badaniami innych właściwych markerów zgodnie z lokalnymi wytycznymi. Pacjenci z dodatnim wynikiem badań serologicznych w kierunku wirusowego zapalenia wątroby typu B (HBsAg lub HBcAb) powinni skonsultować się ze specjalistą chorób wątroby przed rozpoczęciem leczenia oraz powinni być monitorowani i leczeni zgodnie z lokalnymi wytycznymi medycznymi, aby zapobiec reaktywacji zapalenia wątroby typu B.
Leczenie pacjentów z ciężkim osłabieniem odporności
Nie wolno leczyć pacjentów w stanie ciężkiego osłabienia odporności do czasu ustąpienia takiego stanu (patrz punkt 4.3).
Nie zaleca się stosowania innych leków immunosupresyjnych jednocześnie z ofatumumabem, z wyjątkiem kortykosteroidów w objawowym leczeniu rzutów.
Szczepienia
Wszystkie szczepienia należy podać według wytycznych dotyczących immunizacji przynajmniej 4 tygodnie przed rozpoczęciem stosowania ofatumumabu w przypadku szczepionek żywych i żywych atenuowanych oraz, gdy tylko jest to możliwe, przynajmniej 2 tygodnie przed rozpoczęciem podawania ofatumumabu w przypadku szczepionek inaktywowanych.
Ofatumumab może zaburzać skuteczność szczepionek inaktywowanych.
Nie przeprowadzono badań nad bezpieczeństwem immunizacji szczepionkami żywymi ani żywymi atenuowanymi po terapii ofatumumabem. Nie zaleca się podawania szczepionek żywych lub żywych atenuowanych podczas leczenia ani po zakończeniu leczenia do czasu odnowy limfocytów B (patrz punkt 4.5). Mediana czasu do powrotu limfocytów B do dolnej granicy normy (LLN, zdefiniowanej jako 40 komórek / µl) lub wartości wyjściowej wynosi 24,6 tygodnia po zaprzestaniu leczenia, na podstawie danych z badań III fazy (patrz punkt 5.1).
Szczepienie niemowląt, których matki w okresie ciąży leczono ofatumumabem
Niemowlętom, których matki były leczone ofatumumabem w okresie ciąży, nie należy podawać żywych ani żywych atenuowanych szczepionek przed potwierdzeniem u nich powrotu liczby limfocytów B do normy. Deplecja limfocytów B u tych niemowląt może zwiększać ryzyko ze strony szczepionek żywych lub żywych atenuowanych.
Szczepionki inaktywowane można podawać według wskazań przed ustąpieniem deplecji limfocytów B, należy jednak rozważyć ocenę odpowiedzi immunologicznej na szczepienie, w tym konsultację z wykwalifikowanym specjalistą, aby stwierdzić, czy osiągnięto ochronną odpowiedź immunologiczną (patrz punkt 4.6).
Zawartość sodu
Produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, to znaczy lek uznaje się za „wolny od sodu”.
Nie przeprowadzono badań dotyczących interakcji, ponieważ nie są spodziewane interakcje za pośrednictwem enzymów cytochromu P450, innych enzymów metabolizujących leki oraz transporterów.
Szczepienia
Nie badano bezpieczeństwa ani zdolności wywoływania odpowiedzi pierwotnej lub odpowiedzi anamnestycznej (przypominającej) na immunizację szczepionkami żywymi, żywymi atenuowanymi lub inaktywowanymi podczas leczenia ofatumumabem. Odpowiedź na szczepienie przy zmniejszonej liczbie limfocytów B może być osłabiona. Zaleca się, aby pacjenci zakończyli immunizację przed rozpoczęciem terapii ofatumumabem (patrz punkt 4.4).
Inne leki immunosupresyjne lub immunomodulujące
Należy rozważyć ryzyko addytywnego wpływu na układ immunologiczny, gdy leki immunosupresyjne podaje się z ofatumumabem.
Rozpoczynając podawanie ofatumumabu po innych lekach immunosupresyjnych o przedłużonym wpływie na układ immunologiczny lub wdrażając leczenie innymi lekami immunosupresyjnymi o przedłużonym wpływie na układ immunologiczny po ofatumumabie należy wziąć pod uwagę czas trwania i sposób działania tych produktów leczniczych ze względu na potencjalnie addytywne działanie immunosupresyjne (patrz punkt 5.1).
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym powinny stosować skuteczną antykoncepcję (metody, dla których wskaźniki ciąż są mniejsze niż 1%) podczas otrzymywania produktu leczniczego Kesimpta oraz przez 6 miesięcy od ostatniego podania produktu Kesimpta.
Ciąża
Istnieją ograniczone dane dotyczące stosowania ofatumumabu u kobiet w ciąży. Badania na zwierzętach wykazały, że ofatumumab może przenikać przez łożysko i powodować deplecję limfocytów B u płodu (patrz punkt 5.3). Nie obserwowano działania teratogennego po dożylnym podaniu ofatumumabu ciężarnym małpom w okresie organogenezy.
Zgłaszano przypadki przemijającej deplecji limfocytów B we krwi obwodowej i limfocytopenii u niemowląt, których matki były narażone na inne przeciwciała anty-CD20 w okresie ciąży. Potencjalny czas trwania deplecji limfocytów B u niemowląt z ekspozycją na ofatumumab w okresie prenatalnym oraz wpływ deplecji limfocytów B na bezpieczeństwo i skuteczność szczepionek są nieznane (patrz punkty 4.4 i 5.1).
Zaleca się unikanie leczenia ofatumumabem w okresie ciąży, chyba że potencjalne korzyści dla matki przeważają nad ryzykiem dla płodu.
Aby pomóc ustalić wpływ ofatumumabu na kobiety w ciąży zachęca się osoby należące do fachowego personelu medycznego do zgłaszania lokalnemu przedstawicielowi podmiotu odpowiedzialnego wszystkich przypadków ciąży i powikłań, które wystąpią w czasie leczenia lub w ciągu 6 miesięcy od przyjęcia ostatniej dawki ofatumumabu. Umożliwi to monitorowanie tych pacjentek w programie intensywnego monitorowania wpływu na ciążę (ang. PRegnancy outcomes Intensive Monitoring programme, PRIM). Dodatkowo należy zgłaszać wszelkie działania niepożądane związane z ciążą za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
Karmienie piersią
Nie badano stosowania ofatumumabu u kobiet karmiących piersią. Nie wiadomo, czy ofatumumab przenika do mleka ludzkiego. U ludzi przenikanie przeciwciał IgG do mleka występuje w pierwszych kliku dniach po porodzie i niedługo po tym zmniejsza się do niskich stężeń. W rezultacie nie można wykluczyć ryzyka dla dziecka karmionego piersią w tym krótkim okresie. Po tym czasie ofatumumab może być stosowany w okresie karmienia piersią, jeżeli jest to klinicznie uzasadnione. Jeżeli jednak pacjentka była leczona ofatumumabem do ostatnich miesięcy ciąży, karmienie piersią można rozpocząć natychmiast po urodzeniu dziecka.
Płodność
Brak danych dotyczących wpływu ofatumumabu na płodność u ludzi.
Dane niekliniczne nie wskazały ryzyka dla ludzi na podstawie parametrów płodności ocenianych u samic i samców małp.
Produkt leczniczy Kesimpta nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Najważniejszymi i najczęściej zgłaszanymi działaniami niepożądanymi były zakażenia górnych dróg oddechowych (39,4%), ogólnoustrojowe reakcje związane ze wstrzyknięciem (20,6%), reakcje w miejscu wstrzyknięcia (10,9%) oraz zakażenia dróg moczowych (11,9%) (bardziej szczegółowe informacje, patrz punkt 4.4 i część poniżej “Opis wybranych działań niepożądanych”).
Tabelaryczne zestawienie działań niepożądanych
Działania niepożądane zgłaszane w związku ze stosowaniem ofatumumabu w podstawowych badaniach klinicznych nad RMS i w okresie po wprowadzeniu do obrotu wymieniono w Tabeli 1 zgodnie z klasyfikacją układów i narządów MedDRA. W każdej grupie układów i narządów działania niepożądane wymieniono według częstości występowania, zaczynając od najczęstszych. W każdej grupie o określonej częstości działania niepożądane wymieniono według zmniejszającego się nasilenia. Ponadto kategorie częstości dla każdego działania niepożądanego są oparte na następującej konwencji: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1 000 do <1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000) oraz nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 1 Tabelaryczny wykaz działań niepożądanych
|
Zakażenia i zarażenia pasożytnicze |
|
|
Bardzo często |
Zakażenia górnych dróg oddechowych1 |
|
Często |
Opryszczka jamy ustnej |
|
Zaburzenia układu immunologicznego |
|
|
Nieznana |
Reakcje nadwrażliwości |
|
Urazy, zatrucia i powikłania po zabiegach |
|
|
Bardzo często |
Reakcje związane ze wstrzyknięciem (układowe) |
| Zaburzenia żołądka i jelit | |
| Często | Nudności, wymioty4 |
|
Badania diagnostyczne |
|
|
Często |
Zmniejszenie stężenia immunoglobuliny M we krwi |
|
1 Przy określeniu częstości działań niepożądanych użyto preferowany termin zbiorczy (PT) obejmujący: zapalenie błony śluzowej nosa i gardła, zakażenie górnych dróg oddechowych, grypę, zapalenie zatok, zapalenie gardła, zapalenie błony śluzowej nosa, wirusowe zakażenie górnych dróg oddechowych, zapalenie migdałków, ostre zapalenie zatok, zapalenie gardła i migdałków, zapalenie krtani, zapalenie gardła gronkowcowe, wirusowe zapalenie błony śluzowej nosa, bakteryjne zapalenie zatok, zapalenie migdałków bakteryjne, wirusowe zapalenie gardła, wirusowe zapalenie migdałków, przewlekłe zapalenie zatok, opryszczkę nosa, zapalenie tchawicy. 3Zgłaszane w okresie po wprowadzeniu do obrotu (patrz punkt 4.4) 4 Występowanie nudności i wymiotów zgłaszano w związku z ogólnoustrojowymi reakcjami związanymi ze wstrzyknięciem (patrz punkt 4.4). |
|
Opis wybranych działań niepożądanych
Zakażenia
W badaniach klinicznych III fazy nad RMS całkowita częstość występowania zakażeń i poważnych zakażeń u pacjentów leczonych ofatumumabem była podobna, jak u pacjentów leczonych teryflunomidem (odpowiednio 51,6% w por. z 52,7% i 2,5% w por. z 1,8%). Dwóch pacjentów (0,2%) nie kontynuowało a 11 pacjentów (1,2%) przerwało czasowo leczenie w trakcie badania ze względu na poważne zakażenia.
Zakażenia górnych dróg oddechowych
W tych badaniach u 39,4% pacjentów leczonych ofatumumabem wystąpiły zakażenia górnych dróg oddechowych wobec 37,8% pacjentów leczonych teryflunomidem. Zakażenia były przeważnie łagodne do umiarkowanych oraz obejmowały głównie zapalenie nosogardzieli, zakażenie górnych dróg oddechowych i grypę.
Reakcje związane ze wstrzyknięciem
W badaniach klinicznych III fazy nad RMS reakcje związane ze wstrzyknięciem (ogólnoustrojowe) zgłoszono u 20,6% pacjentów leczonych ofatumumabem.
Częstość występowania reakcji związanych ze wstrzyknięciem była największa po pierwszym wstrzyknięciu (14,4%), znamiennie zmniejszając się wraz z kolejnymi wstrzyknięciami (4,4% dla drugiego, <3% od trzeciego wstrzyknięcia). Nasilenie reakcji związanych ze wstrzyknięciem było głównie (99,8%) łagodne do umiarkowanego. Dwóch (0,2%) pacjentów z SM leczonych ofatumumabem zgłosiło poważne, ale niezagrażające życiu reakcje związane ze wstrzyknięciem. Najczęściej zgłaszanymi objawami (≥2%) były gorączka, ból głowy, ból mięśni, dreszcze i zmęczenie.
Dodatkowe zgłaszane objawy obejmowały nudności (1,7%) i wymioty (0,6%).
Reakcje w miejscu wstrzyknięcia
W badaniach klinicznych III fazy nad RMS reakcje w miejscu wstrzyknięcia (miejscowe) zgłoszono u 10,9% pacjentów leczonych ofatumumabem.
Miejscowe reakcje w miejscu podania występowały bardzo często. Reakcje w miejscu wstrzyknięcia były nasilone w stopniu łagodnym do umiarkowanego i nie były to reakcje poważne. Najczęściej zgłaszane objawy (≥2%) obejmowały rumień, ból, swędzenie i obrzęk.
Nieprawidłowe wyniki badań laboratoryjnych
Immunoglobuliny
W przebiegu badań klinicznych III fazy nad RMS obserwowano zmniejszenie średniego stężenia immunoglobuliny M (IgM) (30,9% zmniejszenie po 48 tygodniach i 38,8% po 96 tygodniach) i wykazano brak związku z ryzykiem zakażeń, w tym poważnych zakażeń.
U 14,3% pacjentów leczenie ofatumumabem prowadziło do zmniejszenia stężenia IgM, które osiągnęło wartość poniżej 0,34 g/l.
Stosowanie ofatumumabu było związane z przejściowym zmniejszeniem średniego stężenia immunoglobuliny G (IgG) o 4,3% po 48 tygodniach leczenia, ale ze zwiększeniem o 2,2% po 96 tygodniach.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
W badaniach klinicznych pacjentom z SM podawano dawki do 700 mg bez wystąpienia toksyczności ograniczającej dawkę. W razie przedawkowania zaleca się monitorowanie pacjenta pod kątem podmiotowych i przedmiotowych objawów działań niepożądanych oraz wdrożenie odpowiedniego leczenia objawowego, jeśli zajdzie taka konieczność.
Ofatumumab był wcześniej stosowany w leczeniu przewlekłej białaczki limfocytowej (ang. chronic lymphocytic leukaemia, CLL) w dawkach do 2 000 mg podawanych w infuzji dożylnej. Nie badano ofatumumabu podawanego we wstrzyknięciu podskórnym i nie jest on zatwierdzony do stosowania w tych wskazaniach, a jego stosowanie we wskazaniach onkologicznych nie jest dozwolone.
Grupa farmakoterapeutyczna: leki immunosupresyjne, przeciwciała monoklonalne, kod ATC: L04AG12
Mechanizm działania
Ofatumumab jest w pełni ludzkim przeciwciałem monoklonalnym immunoglobuliną G1 (IgG1) anty-CD20, którego teoretyczna średnia masa cząsteczkowa wynosi 145kDa. Cząsteczka CD20 jest przezbłonową fosfoproteiną z ekspresją na limfocytach B od etapu pre-B po dojrzałe limfocyty B. Ponadto, ekspresję cząsteczki CD20 obserwuje się na niewielkim odsetku aktywowanych limfocytów T. Podskórna droga podania ofatumumabu, a następnie jego uwalnianie/wchłanianie z tkanki umożliwia stopniową interakcję z limfocytami B.
Wiązanie się ofatumumabu z CD20 indukuje lizę limfocytów B z ekspresją CD20, przede wszystkim w mechanizmie cytotoksyczności zależnej od dopełniacza (ang. complement-dependent cytotoxicity, CDC) oraz, w mniejszym stopniu, w mechanizmie cytotoksyczności komórkowej zależnej od przeciwciał (ang. antibody-dependent cell-mediated cytotoxicity, ADCC). Wykazano, że ofatumumab indukuje lizę komórek zarówno z dużą, jak i małą ekspresją CD20. Limfocyty T z ekspresją CD20 są także usuwane przez ofatumumab.
Działanie farmakodynamiczne
Deplecja limfocytów B
W badaniach klinicznych nad RMS podawanie ofatumumabu w dawce 20 mg co 4 tygodnie, po początkowym schemacie dawkowania 20 mg w dniach 1., 7. i 14., spowodowało szybkie i trwałe zmniejszenie liczby limfocytów B do poziomu poniżej DGN (definiowanej jako 40 komórek/µl) już po dwóch tygodniach od rozpoczęcia leczenia. Przed rozpoczęciem fazy leczenia podtrzymującego, począwszy od 4. tygodnia, u 94% pacjentów osiągano całkowitą liczbę limfocytów B <10 komórek/µl, a następnie odsetek ten wzrósł do 98% pacjentów w tygodniu 12. i utrzymywał się przez 120 tygodni (tj. podczas stosowania badanego leczenia).
Powrót liczby limfocytów B do normy
Dane z badań klinicznych III fazy nad RMS wskazują, że mediana czasu do powrotu liczby limfocytów B do DGN lub do wartości wyjściowej wynosi 24,6 tygodnia po zakończeniu leczenia. Te dane zostały potwierdzone w modelowaniu farmakokinetyki limfocytów B i symulacjach powrotu liczby limfocytów B do normy, które przewidują, że mediana czasu do powrotu liczby limfocytów B do DGN wyniesie 23 tygodnie po zakończeniu leczenia.
Immunogenność
W badaniach III fazy nad RMS całkowita częstość występowania przeciwciał przeciwlekowych (ADA) wynosiła 0,2% (2 spośród 914 pacjentów) w grupie pacjentów leczonych ofatumumabem; u żadnego pacjenta nie wykryto przeciwciał przeciwlekowych wzmacniających lub neutralizujących działanie leku. Wpływ dodatniego miana ADA na farmakokinetykę, profil bezpieczeństwa lub kinetykę limfocytów B nie może być oceniony ze względu na niską częstość występowania ADA podczas stosowania ofatumumebu.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność i bezpieczeństwo stosowania ofatumumabu oceniano w dwóch randomizowanych podstawowych badaniach III fazy z podwójnie ślepą próbą, kontrolowanych inną substancją czynną, o jednakowym planie (Badanie 1 [ASCLEPIOS I] i Badanie 2 [ASCLEPIOS II]) z udziałem pacjentów z rzutową postacią SM (RMS) w wieku od 18 do 55 lat, wyjściowym wynikiem niesprawności w rozszerzonej skali niesprawności (ang. Expanded Disability Status Scale, EDSS) od 0 do 5,5 oraz z co najmniej jednym udokumentowanym rzutem choroby w poprzednim roku lub dwoma rzutami w dwóch poprzednich latach, bądź dodatnim wynikiem badania MRI ze wzmocnieniem gadolinem (Gd) w poprzednim roku. Do badań włączono zarówno pacjentów nowo zdiagnozowanych, jak i przestawianych z dotychczasowej terapii.
W dwóch badaniach odpowiednio 927 i 955 pacjentów z RMS przydzielono losowo w stosunku 1:1 do grupy otrzymującej ofatumumab w dawce 20 mg we wstrzyknięciu podskórnym podawanym co 4 tygodnie począwszy od tygodnia 4. po początkowym schemacie podawania trzech dawek 20 mg co tydzień przez pierwszych 14 dni (w dniu 1., 7. i 14.) lub do grupy otrzymującej teryflunomid w dawce 14 mg, w postaci kapsułek przyjmowanych doustnie raz na dobę. Pacjenci otrzymywali także placebo odpowiadające drugiej grupie leczenia, aby zapewnić zaślepienie leczenia (badanie z podwójnie pozorowaną próbą).
Czas trwania leczenia poszczególnych pacjentów był zmienny i zależny od tego, kiedy spełnione zostały kryteria zakończenia badania. W obu badaniach mediana czasu trwania leczenia wyniosła 85 tygodni, a u 33% pacjentów w grupie otrzymującej ofatumumab w porównaniu z 23,2% pacjentów w grupie otrzymującej teryflunomid leczenie trwało ponad 96 tygodni.
Dane demograficzne i charakterystyka wyjściowa były dobrze zrównoważone pomiędzy grupami leczenia i oboma badaniami (patrz Tabela 2). Średni wiek uczestników wynosił 38 lat, średni czas trwania choroby 8,2 roku od wystąpienia pierwszych objawów, a średni wynik w skali EDSS 2,9; 40% pacjentów nie było wcześniej leczonych lekami modyfikującymi przebieg choroby (ang. disease-modifying therapy, DMT), a u 40% wystąpiły zmiany w obrazach T1-zależnych ulegających wzmocnieniu po podaniu gadolinu (Gd) w początkowym badaniu MRI.
Pierwszorzędowym punktem końcowym oceny skuteczności w obu badaniach był roczny odsetek potwierdzonych rzutów choroby (ang. annualised rate of confirmed relapses, ARR) oparty na EDSS. Najważniejsze drugorzędowe punkty końcowe oceny skuteczności obejmowały czas do pogorszenia niesprawności w skali EDSS (potwierdzonego po 3 miesiącach i 6 miesiącach), zdefiniowanego jako wzrost wyniku w skali EDSS o ≥1,5, ≥1 lub ≥0,5 u pacjentów z wyjściowym wynikiem EDSS wynoszącym, odpowiednio, 0, 1 do 5 lub ≥5,5. Dalsze najważniejsze drugorzędowe punkty końcowe obejmowały liczbę zmian w obrazach T1-zależnych ulegających wzmocnieniu po podaniu gadolinu w badaniu MRI, roczny odsetek nowych lub powiększających się zmian w obrazach T2-zależnych, oraz stężenie łańcucha lekkiego neurofilamentu (NfL) w surowicy Najważniejsze drugorzędowe punkty końcowe związane z niesprawnością oceniano w metaanalizie połączonych danych z badań ASCLEPIOS Badanie 1 i Badanie 2, jak zdefiniowano w protokołach badań.
Tabela 2 Dane demograficzne i charakterystyka wyjściowa
|
Charakterystyka |
Badanie 1 |
Badanie 2 |
||
|
|
Ofatumumab |
Teryflunomid |
Ofatumumab |
Teryflunomid |
|
Wiek (średnia ± odchylenie standardowe; lata) |
39±9 |
38±9 |
38±9 |
38±9 |
|
Płeć (kobiety; %) |
68,4 |
68,6 |
66,3 |
67,3 |
|
Czas trwania SM od rozpoznania (średnia/mediana; lata) |
5,77 / 3,94 |
5,64 / 3,49 |
5,59 / 3,15 |
5,48 / 3,10 |
|
Wcześniejsze leczenie DMT (%) |
58,9 |
60,6 |
59,5 |
61,8 |
|
Liczba rzutów choroby w minionych 12 miesiącach |
1,2 |
1,3 |
1,3 |
1,3 |
|
Wynik EDSS (średnia/mediana) |
2,97 / 3,00 |
2,94 / 3,00 |
2,90 / 3,00 |
2,86 / 2,50 |
|
Średnia całkowita objętość zmian w obrazach T2 (cm3) |
13,2 |
13,1 |
14,3 |
12,0 |
|
Pacjenci ze zmianami w obrazach Gd+ T1 (%) |
37,4
|
36,6 |
43,9 |
38,6 |
|
Liczba zmian w obrazach Gd+ T1 (średnia) |
1,7 |
1,2 |
1,6 |
1,5 |
Wyniki dotyczące skuteczności uzyskane w obu badaniach podsumowano w Tabeli 3., na Rycinie 1 oraz na Rycinie 2.
W obu badaniach III fazy ofatumumab w porównaniu z teryflunomidem wykazywał znamienne zmniejszenie rocznego odsetka rzutów, odpowiednio o 50,5% i 58,4%.
Określona a priori metaanaliza połączonych danych wykazała, że ofatumumab porównany z teryflunomidem znamiennie zmniejszał ryzyko 3-miesięcznej potwierdzonej progresji niesprawności (ang. confirmed disability progression, CDP) o 34,3%, a ryzyko 6-miesięcznej CDP o 32,4% (patrz Rycina 1.).
Ofatumumab porównany z teryflunomidem znamiennie zmniejszał liczbę zmian w obrazach T1- zależnych ulegających wzmocnieniu po podaniu gadolinu o 95,9%, a liczbę nowych lub powiększających się zmian w obrazach T2-zależnych o 83,5% (wartości przedstawiają średnie redukcje z połączonych badań).
W porównaniu z teryflunomidem ofatumumab znacząco zmniejszył stężenie NfLjuż od pierwszej oceny po 3 miesiącach (patrz Tabela 3 oraz Rycina 2.).
Podobne działania ofatumumabu na najważniejsze wyniki dotyczące skuteczności w porównaniu z teryflunomidem obserwowano w dwóch badaniach III fazy w podgrupach poddanych analizie eksploracyjnej, wyodrębnionych ze względu na płeć, wiek, masę ciała, wcześniejsze niesteroidowe leczenie SM oraz wyjściowy poziom niesprawności i aktywność choroby.
Tabela 3 Przegląd najważniejszych wyników z badań III fazy nad RMS
|
Punkty końcowe |
Badanie 1 |
Badanie 2 |
||
|
Ofatumumab |
Teryflunomid |
Ofatumumab |
Teryflunomid |
|
|
Punkty końcowe w oparciu o oddzielne badania |
||||
|
Roczny odsetek rzutów (ARR) (pierwszorzędowy punkt końcowy)1 |
0,11 |
0,22 |
0,10 |
0,25 |
|
Redukcja odsetka |
50,5% (p<0,001) |
58,4% (p<0,001) |
||
|
Średnia liczna zmian w obrazach T1- zależnych ulegających wzmocnieniu po Gd w badaniu MRI |
0,0115 |
0,4555 |
0,0317 |
0,5172 |
|
Względna redukcja |
97,5% (p<0,001) |
93,9% (p<0,001) |
||
|
Liczba nowych lub powiększających się zmian w obrazach T2-zależnych na rok |
0,72 |
4,00 |
0,64 |
4,16 |
|
Względna redukcja |
81,9% (p<0,001) |
84,6% (p<0,001) |
||
|
Punkty końcowe w oparciu o predefiniowane metaanalizy |
||||
|
Odsetek pacjentów z 3-miesięczną potwierdzoną progresją niesprawności2 |
10,9% ofatumumab w porównaniu z 15,0% teryflunomid |
|||
|
Odsetek pacjentów z 6-miesięczną potwierdzoną progresją niesprawności2 |
8,1% ofatumumab w porównaniu z 12,0% teryflunomid
|
|||
|
1 Potwierdzone rzuty choroby (z towarzyszącą im klinicznie istotną zmianą w EDSS). |
||||
Rycina 1 Czas do pierwszej 3-miesięcznej CDP według leczenia (połączone dane z Badania 1 i Badania 2 ASCLEPIOS, pełna analizowana grupa)

Rycina 2 Stężenia NfL w surowicy według leczenia (połączone dane z Badania 1 i Badania 2 ASCLEPIOS, pełna analizowana grupa)
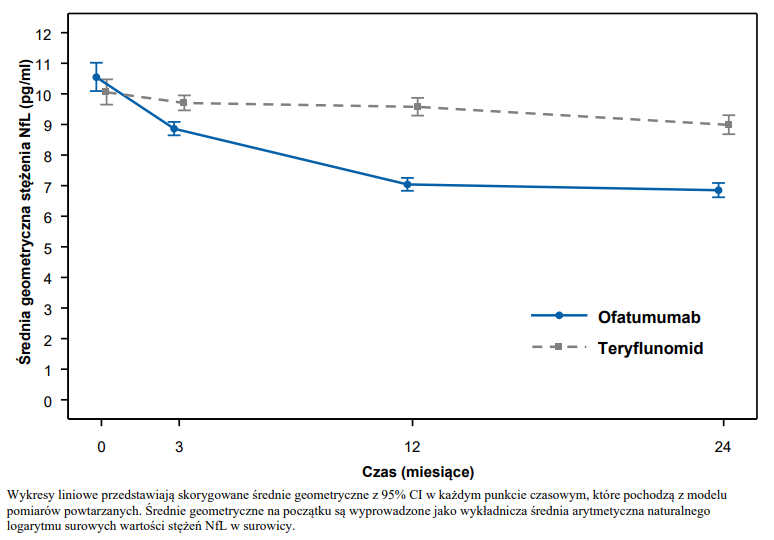
W badaniach III fazy odsetek pacjentów ze zdarzeniami niepożądanymi (ang. adverse events, AE) (83,6% w porównaniu z 84,2%) i AE prowadzącymi do przerwania leczenia (5,7% w porównaniu z 5,2%) były podobne w grupach otrzymujących ofatumumab i teryflunomid.
Dzieci i młodzież
Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego Kesimpta w jednej lub kilku podgrupach populacji dzieci i młodzieży w leczeniu stwardnienia rozsianego (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Wchłanianie
Po podaniu podskórnym ofatumumab charakteryzuje się wydłużonym uwalnianiem/wchłanianiem (Tmax = 4,3 dnia) i jest w przeważającym stopniu wchłaniany przez układ limfatyczny.
Podskórne podawanie dawki 20 mg co miesiąc prowadzi do średniego AUCtau 483 µg*h/ml i średniego Cmax 1,43 µg/ml w stanie stacjonarnym.
Dystrybucja
Objętość dystrybucji w stanie stacjonarnym oszacowano na 5,42 litra po wielokrotnym podaniu ofatumumabu podskórnie w dawce 20 mg.
Metabolizm
Ofatumumab jest białkiem, dla którego spodziewany szlak metaboliczny to rozpad do krótkich peptydów i aminokwasów przy udziale typowych enzymów proteolitycznych.
Eliminacja
Ofatumumab jest usuwany na dwa sposoby: usuwanie w sposób zależny od komórek docelowych, które jest powiązane z wiązaniem się z limfocytami B oraz w sposób niezależny od komórek docelowych, z udziałem nieswoistej endocytozy, a następnie katabolizmu wewnątrzkomórkowego, jak ma to miejsce w przypadku innych cząsteczek IgG. Limfocyty B obecne przed rozpoczęciem leczenia odpowiadają za większy udział klirensu ofatumumabu zależnego od komórek docelowych na początku leczenia. Podawanie ofatumumabu prowadzi do silnej deplecji limfocytów B, co skutkuje zmniejszonym klirensem całkowitym.
Okres półtrwania w stanie stacjonarnym oszacowano na około 16 dni po wielokrotnym podskórnym podaniu ofatumumabu w dawce 20 mg.
Liniowość lub nieliniowość
Farmakokinetyka ofatumumabu jest nieliniowa i związana z klirensem zmniejszającym się wraz z upływem czasu.
Szczególne populacje pacjentów
Dorośli powyżej 55 lat
Nie przeprowadzono specjalnych badań farmakokinetyki ofatumumabu u pacjentów w wieku powyżej 55 lat z powodu ograniczonego doświadczenia klinicznego (patrz punkt 4.2).
Dzieci i młodzież
Nie przeprowadzono badań mających na celu zbadanie farmakokinetyki ofatumumabu u dzieci i młodzieży w wieku poniżej 18 lat.
Płeć
Płeć wywiera niewielki (12%) wpływ na objętość dystrybucji ofatumumabu w kompartmencie centralnym w przekrojowej analizie populacji uczestników badań, z większymi wartościami Cmax i AUC obserwowanymi u kobiet (48% pacjentów w analizie to mężczyźni, a 52% to kobiety); ten wpływ nie jest uznawany za klinicznie istotny i dostosowanie dawki nie jest zalecane.
Masa ciała
Wyniki analizy populacji z kilku badań wskazują, że masa ciała jest współzmienną ekspozycji (Cmax i AUC) na ofatumumab u pacjentów z RMS. Masa ciała nie miała jednak wpływu na miary bezpieczeństwa stosowana i skuteczności oceniane w badaniach klinicznych, dlatego dostosowanie dawki nie jest wymagane.
Zaburzenia czynności nerek
Nie przeprowadzono specjalnych badań ofatumumabu u pacjentów z zaburzeniami czynności nerek.
Pacjenci z łagodnymi zaburzeniami czynności nerek byli włączani do badań klinicznych. Brak doświadczenia u pacjentów z umiarkowanymi i ciężkimi zaburzeniami czynności nerek. Ponieważ jednak ofatumumab nie jest wydalany z moczem, nie należy spodziewać się konieczności modyfikacji dawki u pacjentów z zaburzeniami czynności nerek.
Zaburzenia czynności wątroby
Nie przeprowadzono badań ofatumumabu u pacjentów z zaburzeniami czynności wątroby.
Ponieważ metabolizm wątrobowy przeciwciał monoklonalnych, takich jak ofatumumab, jest nieistotny, nie należy oczekiwać, by zaburzenia czynności wątroby miały wpływ na jego farmakokinetykę. Dlatego nie należy spodziewać się konieczności modyfikacji dawki u pacjentów z zaburzeniami czynności wątroby.
Dane niekliniczne, wynikające z konwencjonalnych badań farmakologicznych dotyczących badań toksyczności po podaniu wielokrotnym, w tym punktów końcowych bezpieczeństwa, nie ujawniają szczególnego zagrożenia dla człowieka.
Nie przeprowadzono badań rakotwórczości i właściwości mutagennych ofatumumabu. Ofatumumab jest przeciwciałem i dlatego nie należy oczekiwać bezpośrednich interakcji ofatumumabu z DNA.
Badania nad wpływem na rozwój zarodka i płodu (EFD) oraz rozszerzone badania rozwoju przed- i pourodzeniowego (ang. enhanced pre/post-natal development, ePPND) u małp wykazały, że ekspozycja na ofatumumab podawany dożylnie w okresie ciąży nie wpływała toksycznie na matkę, nie była też teratogenna ani szkodliwa dla rozwoju zarodka i płodu oraz rozwoju przed- i pourodzeniowego.
W tych badaniach ofatumumab był wykrywany we krwi płodów i młodych, co potwierdzało przenikanie przez łożysko i narażenie płodu na ofatumumab utrzymującą się po urodzeniu (długi okres półtrwania przeciwciał monoklonalnych). Ekspozycja na ofatumumab w okresie ciąży prowadziła do oczekiwanej deplecji limfocytów B z ekspresją CD20 u matek i ich płodów oraz młodych osobników, wraz ze zmniejszeniem masy śledziony (bez korelatów histologicznych) u płodów oraz osłabionej immunologicznej odpowiedzi humoralnej na hemocyjaninę ślimaka morskiego (ang. keyhole limpet haemocyanin, KLH) u młodych osobników (dla wysokich dawek). Wszystkie te zmiany były przemijające w ciągu 6 miesięcy po urodzeniu. U młodych osobników wczesną śmiertelność pourodzeniową zaobserwowano po podaniu dawki 160 razy większej od dawki terapeutycznej (na podstawie AUC) i wynikała ona prawdopodobnie z potencjalnych zakażeń wtórnych do działania immunomodulującego. Wartość NOAEL związana z aktywnością farmakologiczną ofatumumabu u młodych osobników w badaniu ePPND wskazuje na margines bezpieczeństwa na podstawie AUC stanowiącego co najmniej 22-krotność, gdy ekspozycja matki na NOAEL jest porównywalna z ekspozycją u ludzi po podaniu dawki terapeutycznej wynoszącej 20 mg na miesiąc.
W badaniu poświęconym płodności małp nie obserwowano wpływu na punkty końcowe płodności samic i samców.
L-arginina
Sodu octan trójwodny
Sodu chlorek
Polisorbat 80 (E 433)
Disodu edetynian dwuwodny
Kwas solny (do ustalenia odpowiedniego pH)
Woda do wstrzykiwań
Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
3 lata
Kesimpta 20 mg roztwór do wstrzykiwań w ampułko-strzykawce
Przechowywać w lodówce (2°C - 8°C). Nie zamrażać.
W razie potrzeby, produkt leczniczy Kesimpta może być przechowywany poza lodówką jednorazowo przez okres do 7 dni w temperaturze pokojowej (poniżej 30°C). Jeśli produkt leczniczy Kesimpta nie został zużty w tym czasie, może być ponownie przechwywany w lodówce przez okres maksymalnie 7 dni.
Przechowywać ampułko-strzykawkę w opakowaniu zewnętrznym w celu ochrony przed światłem.
Kesimpta 20 mg roztwór do wstrzykiwań we wstrzykiwaczu
Przechowywać w lodówce (2°C - 8°C). Nie zamrażać.
W razie potrzeby, produkt leczniczy Kesimpta może być przechowywany poza lodówką jednorazowo przez okres do 7 dni w temperaturze pokojowej (poniżej 30°C). Jeśli produkt leczniczy Kesimpta nie został zużty w tym czasie, może być ponownie przechwywany w lodówce przez okres maksymalnie 7 dni.
Przechowywać wstrzykiwacz w opakowaniu zewnętrznym w celu ochrony przed światłem.
Kesimpta 20 mg roztwór do wstrzykiwań w ampułko-strzykawce
Produkt leczniczy Kesimpta jest dostarczany w szklanej strzykawce jednorazowego użytku, wyposażonej w igłę ze stali nierdzewnej, uszczelkę tłoka oraz sztywną osłonę igły. Strzykawka ma zamontowany tłok i zabezpieczenie igły.
Produkt leczniczy Kesimpta jest dostępny w opakowaniach jednostkowych zawierających 1 ampułkostrzykawkę oraz w opakowaniach zbiorczych zawierających 3 (3 opakowania po 1) ampułkostrzykawki.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Kesimpta 20 mg roztwór do wstrzykiwań we wstrzykiwaczu
Produkt Kesimpta jest dostarczany w szklanej strzykawce jednorazowego użytku, wyposażonej w igłę ze stali nierdzewnej, uszczelkę tłoka oraz sztywną osłonę igły. Strzykawka jest zamontowana w autowstrzykiwaczu.
Produkt Kesimpta jest dostępny w opakowaniach jednostkowych zawierających 1 wstrzykiwacz półautomatyczny napełniony oraz w opakowaniach zbiorczych zawierających 3 (3 opakowania po 1) wstrzykiwacze półautomatyczne napełnione.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Instrukcja przygotowania ampułko-strzykawki
Przed wstrzyknięciem ampułko-strzykawkę należy wyjąć z lodówki na około 15 do 30 minut, aby lek osiągnął temperaturę pokojową. Ampułko-strzykawkę należy przechowywać w oryginalnym opakowaniu do czasu użycia, a nasadki igły nie należy zdejmować, aż do momentu wykonywania wstrzyknięcia. Przed użyciem roztwór należy ocenić wzrokowo przez okienko. Ampułko-strzykawki nie wolno używać, jeżeli płyn zawiera widoczne cząstki lub jest mętny.
Szczegółowe instrukcje podania produktu zamieszczono w ulotce dla pacjenta.
Instrukcja przygotowania wstrzykiwacza półautomatycznego napełnionego
Przed wstrzyknięciem wstrzykiwacz należy wyjąć z lodówki na około 15 do 30 minut, aby lek osiągnął temperaturę pokojową. Wstrzykiwacz należy przechowywać w oryginalnym opakowaniu do czasu użycia, a nasadki nie należy zdejmować aż do momentu wykonywania wstrzyknięcia. Przed użyciem roztwór należy ocenić wzrokowo przez okienko. Wstrzykiwacza nie wolno używać, jeżeli płyn zawiera widoczne cząstki lub jest mętny.
Szczegółowe instrukcje podania produktu zamieszczono w ulotce dla pacjenta.
Usuwanie
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Novartis Ireland Limited
Vista Building
Elm Park, Merrion Road
Ballsbridge
Dublin 4
Irlandia
EU/1/21/1532/001-004
26 marca 2021
Szczegółowe informacje o tym produkcie leczniczym są dostępne na stronie internetowej Europejskiej Agencji Leków https://www.ema.europa.eu.
Powered by Biogen Poland Sp. z o.o.
(Biogen-101710)
 i
i  "Dodaj aplikację do ekranu początkowego"
"Dodaj aplikację do ekranu początkowego"