Powered by Biogen Poland Sp. z o.o.
Powered by Biogen Poland Sp. z o.o.
Mayzent 0,25 mg tabletki powlekane
Mayzent 1 mg tabletki powlekane
Mayzent 2 mg tabletki powlekane
Mayzent 0,25 mg tabletki powlekane
Każda tabletka powlekana zawiera siponimod z kwasem fumarowym w ilości odpowiadającej 0,25 mg siponimodu.
Substancje pomocnicze o znanym działaniu
Każda tabletka zawiera 59,1 mg laktozy (w postaci jednowodnej) i 0,092 mg lecytyny sojowej.
Mayzent 1 mg tabletki powlekane
Każda tabletka powlekana zawiera siponimod z kwasem fumarowym w ilości odpowiadającej 1 mg siponimodu.
Substancje pomocnicze o znanym działaniu
Każda tabletka zawiera 58,3 mg laktozy (w postaci jednowodnej) i 0,092 mg lecytyny sojowej.
Mayzent 2 mg tabletki powlekane
Każda tabletka powlekana zawiera siponimod z kwasem fumarowym w ilości odpowiadającej 2 mg siponimodu.
Substancje pomocnicze o znanym działaniu
Każda tabletka zawiera 57,3 mg laktozy (w postaci jednowodnej) i 0,092 mg lecytyny sojowej. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana
Mayzent 0,25 mg tabletki powlekane
Bladoczerwone, okrągłe, obustronnie wypukłe tabletki powlekane o ściętych brzegach, o średnicy około 6,1 mm z logo firmy po jednej stronie i symbolem “T” po drugiej stronie tabletki.
Mayzent 1 mg tabletki powlekane
Fioletowo-białe, okrągłe, obustronnie wypukłe tabletki powlekane o ściętych brzegach, o średnicy około 6,1 mm z logo firmy po jednej stronie i symbolem “L” po drugiej stronie tabletki.
Mayzent 2 mg tabletki powlekane
Bladożółte, okrągłe, obustronnie wypukłe tabletki powlekane o ściętych brzegach, o średnicy około 6,1 mm z logo firmy po jednej stronie i symbolem “II” po drugiej stronie tabletki.
Produkt leczniczy Mayzent jest wskazany w leczeniu dorosłych pacjentów z wtórnie postępującą postacią stwardnienia rozsianego (ang. secondary progressive multiple sclerosis, SPMS) z aktywnością choroby potwierdzoną występowaniem nawrotów lub cechami aktywności zapalnej w badaniach obrazowych (patrz punkt 5.1).
Leczenie siponimodem powinno być rozpoczynane i nadzorowane przez lekarza posiadającego doświadczenie w leczeniu pacjentów ze stwardnieniem rozsianym.
Przed rozpoczęciem leczenia u pacjentów należy koniecznie wykonać badanie genotypu CYP2C9, aby określić u nich metabolizm leku przy udziale CYP2C9 (patrz punkty 4.4, 4.5 i 5.2). U pacjentów z genotypem CYP2C9*3*3 nie należy stosować siponimodu (patrz punkty 4.3, 4.4 i 5.2).
Dawkowanie
Rozpoczęcie leczenia
Leczenie należy rozpoczynać od opakowania przeznaczonego do zwiększania dawki, które wystarcza na 5 dni. Leczenie rozpoczyna się od dawki 0,25 mg przyjmowanej raz na dobę w 1. i 2. dniu, a następnie pacjent przyjmuje raz na dobę dawkę 0,5 mg w 3. dniu, dawkę 0,75 mg w 4. dniu i dawkę 1,25 mg w 5. dniu tak, by w 6. dniu osiągnąć przepisaną przez lekarza dawkę podtrzymującą siponimodu (patrz tabela 1).
Podczas pierwszych 6 dni od rozpoczęcia leczenia zalecaną dawkę dobową należy przyjmować raz na dobę rano, z posiłkiem lub bez posiłku.
Tabela 1 Schemat stopniowego zwiększania dawki aż do osiągnięcia dawki podtrzymującej
Zwiększenie dawki
|
Dawka, jaką należy przyjąć
|
Schemat zwiększania dawki
|
Nazwa dawki
|
Dzień 1. Dzień 2. Dzień 3. Dzień 4. Dzień 5. |
0,25 mg 0,25 mg 0,5 mg 0,75 mg 1,25 mg |
1 x 0,25 mg 1 x 0,25 mg 2 x 0,25 mg 3 x 0,25 mg 5 x 0,25 mg |
DAWKA STOPNIOWO ZWIĘKSZANA
|
Dzień 6.
|
2 mg1
|
1 x 2 mg1
|
DAWKA PODTRZYMUJĄCA
|
1U pacjentów z genotypem CYP2C9*2*3 lub *1*3 zalecana dawka podtrzymująca wynosi 1 mg raz na dobę (1 x 1 mg lub 4 x 0,25 mg) (patrz wyżej oraz punkty 4.4 i 5.2). Dodatkowa ekspozycja wynosząca 0,25 mg w dniu 5. nie ma wpływu na bezpieczeństwo pacjenta.
|
|||
Leczenie podtrzymujące
U pacjentów z genotypem CYP2C9*2*3 lub *1*3 zalecana dawka podtrzymująca wynosi 1 mg (patrz punkty 4.4 i 5.2).
Zalecana dawka podtrzymująca siponimodu u pacjentów z wszystkimi innymi genotypami CYP2C9 wynosi 2 mg.
Produkt leczniczy Mayzent należy przyjmować raz na dobę.
Pominięcie dawki (dawek) podczas rozpoczynania leczenia
Jeśli w ciągu pierwszych 6 dni leczenia jednego dnia dojdzie do pominięcia stopniowo zwiększanej dawki, leczenie należy rozpocząć ponownie korzystając z nowego opakowania przeznaczonego do zwiększana dawki.
Pominięcie dawki po 6. dniu leczenia
W razie pominięcia dawki należy przyjąć przepisaną dawkę w kolejnym wyznaczonym terminie; nie należy podwajać kolejnej dawki.
Ponowne rozpoczęcie leczenia podtrzymującego po przerwaniu leczenia
Jeśli leczenie podtrzymujące zostanie przerwane na 4 lub więcej kolejnych dawek dobowych, leczenie siponimodem należy ponownie rozpocząć korzystając z nowego opakowania przeznaczonego do zwiększania dawki.
Szczególne populacje pacjentów
Pacjenci w podeszłym wieku
Siponimod nie był badany u pacjentów w wieku 65 lat i starszych. W badaniach klinicznych uczestniczyli pacjenci w wieku do 61 lat. Siponimod należy stosować z zachowaniem ostrożności u pacjentów w podeszłym wieku z powodu niewystarczających danych dotyczących bezpieczeństwa stosowania i skuteczności (patrz punkt 5.2).
Zaburzenia czynności nerek
Farmakologiczne badania kliniczne wskazują, że nie jest konieczne dostosowanie dawki u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2).
Zaburzenia czynności wątroby
Siponimodu nie wolno stosować u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasy C w skali Child-Pugh) (patrz punkt 4.3). Chociaż nie jest konieczne dostosowanie dawki u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności wątroby, należy zachować ostrożność rozpoczynając leczenie u tych pacjentów (patrz punkty 4.4 i 5.2).
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności siponimodu u dzieci i młodzieży w wieku od 0 do 18 lat. Dane nie są dostępne.
Sposób podawania
Podanie doustne. Siponimod przyjmuje się z pokarmem lub bez pokarmu.
Tabletki powlekane należy połykać w całości popijając wodą.
Zakażenia
Ryzyko zakażeń
Głównym działaniem farmakodynamicznym siponimodu jest zależne od dawki zmniejszenie liczby limfocytów obwodowych do 20-30% wartości wyjściowych. Działanie to jest wynikiem odwracalnego zatrzymania limfocytów w tkankach limfatycznych (patrz punkt 5.1).
Działanie siponimodu na układ immunologiczny może zwiększać ryzyko zakażeń (patrz punkt 4.8).
Przed rozpoczęciem leczenia powinny być dostępne aktualne (tj. wykonane w ciągu ostatnich 6 miesięcy lub po zakończeniu wcześniejszego leczenia) wyniki badania pełnej morfologii krwi. Ocena morfologii krwi jest zalecana również po 3-4 miesiącach od rozpoczęcia leczenia, a później co najmniej raz w roku i w przypadku wystąpienia objawów zakażenia. Potwierdzona, bezwzględna liczba limfocytów <0,2 x 109 /l powinna prowadzić do zmniejszenia dawki do 1 mg, ponieważ w badaniach klinicznych dawkę siponimodu zmniejszano u pacjentów z bezwzględną liczbą limfocytów <0,2 x 109 /l. Potwierdzona, bezwzględna liczba limfocytów <0,2 x 109 /l u pacjenta już stosującego siponimod w dawce 1 mg powinna prowadzić do przerwania leczenia siponimodem do czasu osiągnięcia wartości 0,6 x 109 /l i wówczas można rozważyć wznowienie leczenia siponimodem.
Rozpoczęcie leczenia należy odroczyć u pacjentów z ciężkim czynnym zakażeniem, aż do jego ustąpienia. Ponieważ resztkowe działanie farmakologiczne, takie jak zmniejszenie liczby limfocytów obwodowych może utrzymywać się do 3-4 tygodni po zakończeniu leczenia, w tym okresie należy nadal prowadzić czujną obserwację pacjenta pod kątem zakażeń (patrz punkt „Zakończenie leczenia siponimodem” poniżej).
Należy pouczyć pacjentów, aby niezwłocznie zgłaszali objawy zakażenia lekarzowi prowadzącemu. U pacjentów z objawami zakażenia podczas terapii należy stosować skuteczne strategie rozpoznania i leczenia. Należy rozważyć wstrzymanie leczenia siponimodem w przypadku wystąpienia ciężkiego zakażenia.
Po zastosowaniu siponimodu zgłoszono przypadki kryptokokowego zapalenia opon mózgowych (CM). U pacjentów z przedmiotowymi i podmiotowymi objawami odpowiadającymi CM należy szybko przeprowadzić badania diagnostyczne. Leczenie siponimodem należy wstrzymać do czasu wykluczenia CM. W przypadku rozpoznania CM należy rozpocząć odpowiednie leczenie.
Postępująca wieloogniskowa leukoencefalopatia
Zgłaszano przypadki postępującej wieloogniskowej leukoencefalopatii (ang. progressive multifocal leukoencephalopathy, PML) po zastosowaniu siponimodu (patrz punkt 4.8). Lekarze powinni zachować czujność w odniesieniu do objawów klinicznych lub wyników obrazowania metodą rezonansu magnetycznego (MRI), które mogą sugerować PML. W przypadku podejrzenia PML leczenie siponimodem należy wstrzymać do czasu wykluczenia PML. Jeśli potwierdzono PML, leczenie siponimodem należy przerwać.
U pacjentów leczonych modulatorami receptorów sfingozyno-1-fosforanu (ang. sphingosine 1-phosphate, S1P), w tym siponimodem, u których wystąpiła PML i którzy następnie zakończyli leczenie, zgłaszano występowanie zapalnego zespołu rekonstrukcji immunologicznej (ang. immune reconstitution inflammatory syndrome, IRIS). IRIS objawia się pogorszeniem stanu klinicznego pacjenta, które może być szybkie, prowadzić do ciężkich powikłań neurologicznych lub zgonu i często jest związane z występowaniem charakterystycznych zmian w obrazie MRI. Czas do wystąpienia IRIS u pacjentów z PML zazwyczaj wynosił od kilku tygodni do kilku miesięcy po odstawieniu modulatora receptorów S1P. Należy monitorować pacjentów pod kątem rozwoju IRIS i wdrożyć odpowiednie leczenie powiązanego z nim stanu zapalnego.
Zakażenie wirusem opryszczki
Po zastosowaniu siponimodu w dowolnym momencie podczas leczenia występowały przypadki zakażenia wirusem opryszczki (w tym przypadki zapalenia opon mózgowych lub zapalenia opon mózgowych i mózgu spowodowane przez wirusy ospy wietrznej i półpaśca [VZV]). Jeśli wystąpi opryszczkowe zapalenie opon mózgowych lub opon mózgowych i mózgu, należy przerwać stosowanie siponimodu i zastosować odpowiednie leczenie zakażenia. Pacjenci bez przebytej ospy wietrznej potwierdzonej przez lekarza lub bez udokumentowanego pełnego kursu szczepienia przeciwko wirusowi VZV powinni zostać poddani badaniu na obecność przeciwciał przeciwko VZV przed rozpoczęciem leczenia siponimodem (patrz punkt „Szczepienia” poniżej).
Szczepienia
Przed rozpoczęciem leczenia siponimodem u pacjentów z ujemnym wynikiem badania na obecność przeciwciał zaleca się pełny cykl szczepień przeciwko ospie, po którym należy odczekać 1 miesiąc zanim rozpocznie się leczenie, aby wystąpił pełny efekt szczepienia (patrz punkt 4.8).
Należy unikać stosowania żywych szczepionek atenuowanych podczas przyjmowania siponimodu i przez 4 tygodnie po zakończeniu leczenia (patrz punkt 4.5).
Inne rodzaje szczepionek mogą być mniej skuteczne, jeśli zostaną podane podczas leczenia siponimodem (patrz punkt 4.5). Zaleca się przerwanie leczenia 1 tydzień przed planowanym szczepieniem i nie wznawianie go do 4 tygodni po szczepieniu. Jeśli leczenie siponimodem jest przerwane z powodu szczepienia, należy wziąć pod uwagę możliwy nawrót aktywności choroby (patrz poniżej punkt „Przerwanie leczenia siponimodem”).
Jednoczesne leczenie przeciwnowotworowe, immunomodulujące lub immunosupresyjne
Należy zachować ostrożność podając jednocześnie leki przeciwnowotworowe, immunomodulujące lub immunosupresyjne (w tym kortykosteroidy) ze względu na ryzyko addycyjnego działania na układ immunologiczny podczas takiej terapii (patrz punkt 4.5).
Obrzęk plamki żółtej
W badaniu klinicznym III fazy obrzęk plamki żółtej w połączeniu z objawami ocznymi lub bez tych objawów był częściej zgłaszany po zastosowaniu siponimodu (1,8%) niż placebo (0,2%) (patrz punkt 4.8). Większość przypadków występowała w ciągu pierwszych 3-4 miesięcy leczenia. Z tego względu po 3-4 miesiącach od rozpoczęcia leczenia zaleca się wykonanie badania okulistycznego. Ponieważ przypadki obrzęku plamki żółtej występowały również podczas długotrwałego leczenia, pacjenci powinni zgłaszać zaburzenia widzenia występujące w dowolnym momencie podczas leczenia siponimodem i zaleca się ocenę dna oka obejmującą badanie plamki żółtej.
Nie należy rozpoczynać leczenia siponimodem u pacjentów z obrzękiem plamki żółtej aż do jego ustąpienia.
Siponimod należy stosować z zachowaniem ostrożności u pacjentów z cukrzycą, zapaleniem błony naczyniowej w wywiadzie lub występującą wcześniej/współwystępującą chorobą siatkówki, ze względu na potencjalne zwiększenie ryzyka obrzęku plamki żółtej (patrz punkt 4.8). Zaleca się, by u tych pacjentów wykonywać badanie okulistyczne przed rozpoczęciem leczenia i regularnie podczas stosowania siponimodu, aby wykryć obrzęk plamki żółtej.
Nie oceniano skutków kontynuacji leczenia siponimodem u pacjentów z obrzękiem plamki żółtej. Zaleca się przerwanie leczenia siponimodem, jeśli u pacjenta wystąpi obrzęk plamki żółtej. Przed podjęciem decyzji o ewentualnym wznowieniu leczenia siponimodem po ustąpieniu obrzęku plamki żółtej, należy wziąć pod uwagę potencjalne korzyści i ryzyko u konkretnego pacjenta.
Bradyarytmia
Rozpoczęcie leczenia siponimodem powoduje przemijające zmniejszenie częstości akcji serca i może także wiązać się z opóźnieniem przewodzenia przesionkowo- komorowego (patrz punkty 4.8 i 5.1) . Dlatego na początku leczenia stosuje się schemat stopniowego zwiększania dawki umożliwiający osiągnięcie dawki podtrzymującej w dniu 6. (patrz punkt 4.2).
Po podaniu pierwszej dawki w schemacie jej stopniowego zwiększania akcja serca ulega spowolnieniu w ciągu jednej godziny, a maksymalne zmniejszenie częstości akcji serca w dniu 1. następuje po około 3-4 godzinach. Podczas dalszego zwiększania dawki spowolnienie akcji serca utrzymuje się w kolejnych dniach, przy czym maksymalne zmniejszenie częstości akcji serca od dnia 1. (wartość początkowa) osiągane jest od 5. do 6. dnia leczenia. Największe dobowe zmniejszenie średniej częstości akcji serca mierzonej co godzinę, podane w wartościach bezwzględnych obserwuje się w dniu 1., ze średnim zmniejszeniem tętna o 5 do 6 uderzeń na minutę. Zmniejszenie częstości akcji serca po podaniu dawki leku w kolejnych dniach jest mniej wyraźne. Podczas dalszego nieprzerwanego podawania leku częstość akcji serca zaczyna zwiększać się po dniu 6. i osiąga wartości takie same, jak w grupie otrzymującej placebo w ciągu 10 dni od rozpoczęcia leczenia.
Częstość akcji serca poniżej 40 uderzeń na minutę była rzadko obserwowana.
W większości przypadków opóźnienia przewodzenia przedsionkowo-komorowego objawiały się jako bloki przedsionkowo-komorowe (AV) pierwszego stopnia (wydłużenie odstępu PR w zapisie elektrokardiograficznym). W badaniach klinicznych bloki przedsionkowo-komorowe drugiego stopnia, zazwyczaj typu Mobitz I (Wenckebacha) obserwowano u mniej niż 1,7% pacjentów w chwili rozpoczynania leczenia. Większośc zdarzeń bradyarytmii lub opóźnień przewodzenia przedsionkowo- komorowego była bezobjawowa, przemijająca, ustępowała w ciągu 24 godzin i nie wymagały przerwania leczenia. W przypadku wystąpienia objawów po podaniu dawki (zawroty głowy, ból w klatce piersiowej niezwiązany z sercem i ból głowy), należy wdrożyć odpowiednie postępowanie kliniczne i kontynuować monitorowanie pacjenta do czasu ustąpienia objawów. W razie konieczności zmniejszenie częstości akcji serca wywołane przez siponimod można odwrócić podając pozajelitowe dawki atropiny lub izoprenaliny.
Zalecenia dotyczące rozpoczynania leczenia u pacjentów z pewnymi występującymi wcześniej chorobami serca
W ramach środków ostrożności pacjenci z następującymi chorobami serca powinni być poddani obserwacji przez 6 godzin po podaniu pierwszej dawki siponimodu w celu wykrycia przedmiotowych i podmiotowych objawów bradykardii (patrz także punkt 4.3):
U tych pacjentów zaleca się, by przed podaniem dawki i pod koniec okresu obserwacji wykonać badanie elektrokardiograficzne (EKG). Jeśli po podaniu dawki wystąpi bradyarytmia lub objawy związane z zaburzeniami przewodzenia lub jeśli badanie EKG wykonane po 6 godzinach od podania dawki wykaże nowy blok przedsionkowo-komorowy drugiego lub wyższego stopnia lub QTc ≥500 ms, należy rozpocząć odpowiednie postępowanie i kontynuować obserwację pacjenta aż do ustąpienia tych objawów/wyników. Jeśli konieczne jest leczenie farmakologiczne, należy kontynuować monitorowanie pacjenta do następnego dnia i powtórzyć 6-godzinne monitorowanie po podaniu drugiej dawki.
Ze względu na ryzyko poważnych zaburzeń rytmu serca lub istotnej bradykardii, siponimodu nie należy stosować u pacjentów z:
U tych pacjentów leczenie siponimodem należy rozważyć wyłącznie, jeśli przewidywane korzyści przewyższają potencjalne zagrożenia oraz należy skonsultować się z kardiologiem przed rozpoczęciem leczenia, aby ustalić najbardziej odpowiednią strategię monitorowania stanu pacjenta. Wnikliwe badanie QT wykazało brak istotnego bezpośredniego wpływu siponimodu na wydłużenie odstępu QT oraz brak potencjalnego arytmogennego działania siponimodu związanego z wydłużeniem odstępu QT. Rozpoczęcie leczenia może spowodować zmniejszenie częstości akcji serca i pośrednie wydłużenie odstępu QT w fazie stopniowego zwiększania dawki. Stosowanie siponimodu nie było badane u pacjentów z istotnym wydłużeniem odstępu QT (QTc >500 ms) lub u pacjentów leczonych produktami leczniczymi wydłużającymi odstęp QT. Jeśli leczenie siponimodem jest rozważane u pacjentów z wcześniej występującym istotnym wydłużeniem odstępu QT lub u pacjentów już leczonych produktami leczniczymi wydłużającymi odstęp QT o znanych właściwościach arytmogennych, należy skonsultować się z kardiologiem przed rozpoczęciem leczenia, aby określić najbardziej odpowiednią strategię monitorowania stanu pacjenta podczas rozpoczynania leczenia.
Stosowanie siponimodu nie było badane u pacjentów z zaburzeniami rytmu serca wymagającymi leczenia lekami przeciwarytmicznymi klasy Ia (np. chinidyną, prokainamidem) lub klasy III (np. amiodaronem, sotalolem). Podawanie leków przeciwarytmicznych klasy Ia i klasy III było związane z występowaniem przypadków torsade de pointes u pacjentów z bradykardią. Ponieważ rozpoczęcie leczenia powoduje zmniejszenie częstości akcji serca, siponimodu nie należy stosować jednocześnie z tymi produktami leczniczymi podczas rozpoczynania leczenia.
Doświadczenie ze stosowaniem siponimodu jest ograniczone u pacjentów otrzymujących jednocześnie leki blokujące kanały wapniowe spowalniające czynność serca (takie jak werapamil lub diltiazem) lub inne substancje mogące zmniejszać częstość akcji serca (np. iwabradyna lub digoksyna), ponieważ te produkty lecznicze nie były badane u pacjentów otrzymujących siponimod w badaniach klinicznych. Jednoczesne stosowanie tych substancji podczas rozpoczynania leczenia może wiązać się z wystąpieniem ciężkiej bradykardii i bloku serca. Z uwagi na możliwe działanie addycyjne na częstość akcji serca na ogół nie należy rozpoczynać leczenia siponimodem u pacjentów przyjmujących jednocześnie te substancje (patrz punkt 4.5). U tych pacjentów leczenie siponimodem należy rozważać wyłącznie, jeśli przewidywane korzyści przewyższają potencjalne zagrożenia.
Jeśli podczas rozpoczynania leczenia siponimodem rozważa się jednoczesne leczenie jedną z wyżej wymienionych substancji, należy skonsultować się z kardiologiem odnośnie zmiany stosowanej terapii na leczenie produktami leczniczymi niepowodującymi zmniejszenia częstości akcji serca lub odpowiedniego monitorowania podczas rozpoczynania leczenia.
Działanie bradyarytmiczne jest bardziej nasilone, gdy siponimod jest dodawany do terapii lekami beta-adrenolitycznymi. U pacjentów otrzymujących lek beta-adrenolityczny w stałej dawce, przed rozpoczęciem leczenia należy uwzględnić częstość akcji serca w spoczynku. Jeśli częstość akcji serca w spoczynku wynosi >50 uderzeń na minutę w trakcie przewlekłego leczenia lekiem beta-adrenolitycznym, można rozpocząć leczenie siponimodem. Jeśli częstość akcji serca w spoczynku wynosi ≤50 uderzeń na minutę, wówczas leczenie lekiem beta-adrenolitycznym należy przerwać aż do czasu, gdy wyjściowa częstość akcji serca wyniesie >50 uderzeń na minutę. Można wówczas rozpocząć leczenie siponimodem, a leczenie lekiem beta-adrenolitycznym można wznowić po tym, jak dawka siponimodu zostanie zwiększona do docelowej dawki podtrzymującej (patrz punkt 4.5).
Czynność wątroby
Przed rozpoczęciem leczenia siponimodem należy zapoznać się z ostatnimi (tj. wykonanymi w ciągu ostatnich 6 miesięcy) wynikami badań aktywności aminotransferaz i stężenia bilirubiny.
W badaniu klinicznym III fazy aktywność aminotransferazy alaninowej (AlAT) lub aminotransferazy asparaginianowej (AspAT) wynoszącą trzykrotność górnej granicy normy (GGN) obserwowano u 5,6% pacjentów leczonych siponimodem w dawce 2 mg w porównaniu z 1,5% pacjentów otrzymujących placebo (patrz punkt 4.8). W badaniach klinicznych leczenie przerywano, jeśli zwiększenie to przekraczało 3-krotny wzrost, a u pacjenta występowały objawy związane z zaburzeniami czynności wątroby lub jeśli zwiększenie to przekraczało 5-krotny wzrost. W badaniu klinicznym III fazy 1% wszystkich przypadków przerwania leczenia spełniał jedno z tych kryteriów.
U pacjentów, u których podczas leczenia wystąpią objawy wskazujące na zaburzenia czynności wątroby, należy zbadać aktywność enzymów wątrobowych i przerwać leczenie siponimodem, jeśli potwierdzone będzie istotne uszkodzenie wątroby. Wznowienie terapii będzie zależało od stwierdzenia innej przyczyny uszkodzenia wątroby oraz od korzyści dla pacjenta wynikających ze wznowienia leczenia w porównaniu z ryzykiem nawrotu zaburzeń czynności wątroby.
Mimo że nie ma danych pozwalających stwierdzić, że u pacjentów z wcześniej występującą chorobą wątroby istnieje zwiększone ryzyko wystąpienia podwyższonych wartości wyników testów czynnościowych wątroby podczas przyjmowania siponimodu, należy zachować ostrożność u pacjentów z istotną chorobą wątroby w wywiadzie.
Nowotwory skóry
U pacjentów otrzymujących siponimod, a zwłaszcza u pacjentów poddanych długotrwałemu leczeniu zgłaszano występowania raka podstawnokomórkowego (ang. basal cell carcinoma, BCC) i innych nowotworów skóry, w tym raka kolczystokomórkowego (ang. squamous cell carcinoma, SCC)oraz czerniaka złośliwego (patrz punkt 4.8).
U wszystkich pacjentów zaleca się przeprowadzenie badania skóry w chwili rozpoczynania leczenia, a następnie co 6 do 12 miesięcy biorąc pod uwagę ocenę kliniczną. Podczas dłuższego leczenia należy utrzymać wymóg dokładnego badania skóry. Należy doradzić pacjentom, by niezwłocznie zgłaszali lekarzowi prowadzącemu wszelkie podejrzane zmiany skórne. Pacjentów leczonych siponimodem należy przestrzec przed ekspozycją na światło słoneczne bez ochrony skóry. Pacjenci ci nie powinni jednocześnie otrzymywać fototerapii promieniowaniem UVB lub fotochemioterapii PUVA.
Nieoczekiwane neurologiczne lub psychiczne objawy podmiotowe/przedmiotowe
Po zastosowaniu innego modulatora receptora sfingozyno-1-fosforanu (S1P) zgłaszano rzadkie przypadki zespołu odwracalnej tylnej encefalopatii (PRES). Zdarzeń takich nie zgłaszano po zastosowaniu siponimodu w programie rozwoju leku. Jeśli jednak u pacjenta leczonego siponimodem wystąpią jakiekolwiek nieoczekiwane podmiotowe/przedmiotowe objawy neurologiczne lub psychiczne (np. deficyty poznawcze, zmiany zachowania, korowe zaburzenia widzenia lub wszelkie inne neurologiczne korowe objawy podmiotowe/przedmiotowe lub wszelkie podmiotowe/przedmiotowe objawy wskazujące na zwiększenie ciśnienia śródczaszkowego) bądź przyspieszenie pogarszania się stanu neurologicznego, należy niezwłocznie przeprowadzić u pacjenta pełne badanie fizykalne i neurologiczne oraz rozważyć wykonanie badania MRI.
Wcześniejsze leczenie lekami immunosupresyjnymi lub immunomodulującymi
Zmieniając leczenie z innego leku modyfikującego przebieg choroby, należy uwzględnić jego okres półtrwania i sposób działania, aby uniknąć addycyjnego wpływu na układ immunologiczny, ale również zminimalizować ryzyko reaktywacji choroby. Przed rozpoczęciem leczenia siponimodem zaleca się wykonanie pełnego badania krwi pozwalającego stwierdzić ustąpienie działań dotychczas stosowanych leków na układ immunologiczny pacjenta (tj. cytopenii).
Ze względu na charakterystykę i czas trwania immunosupresyjnych działań alemtuzumabu opisanych w informacji o tym produkcie, nie zaleca się rozpoczynania leczenia siponimodem po leczeniu alemtuzumabem.
Leczenie siponimodem można na ogół rozpocząć bezpośrednio po zakończeniu leczenia interferonem beta lub octanem glatirameru.
Wpływ na ciśnienie krwi
Pacjenci z nadciśnieniem niekontrolowanym za pomocą produktów leczniczych byli wykluczeni z badań klinicznych; u pacjentów z niekontrolowanym nadciśnieniem wskazane jest zachowanie szczególnej ostrożności podczas leczenia siponimodem.
W badaniu klinicznym III fazy z udziałem pacjentów z SPMS nadciśnienie tętnicze zgłaszano częściej u pacjentów leczonych siponimodem (12,6%) niż u pacjentów otrzymujących placebo (9,0%). Leczenie siponimodem spowodowało zwiększenie skurczowego i rozkurczowego ciśnienia krwi. a działanie to rozpoczynało się wcześnie po rozpoczęciu leczenia, osiągając maksymalne nasilenie po około 6 miesiącach leczenia (ciśnienie skurczowe 3 mmHg, ciśnienie rozkurczowe 1,2 mmHg), a następnie pozostając na stabilnym poziomie. Działanie to utrzymywało się w miarę kontynuowania leczenia.
Podczas leczenia siponimodem należy regularnie kontrolować ciśnienie krwi.
Genotyp CYP2C9
Przed rozpoczęciem leczenia siponimodem u pacjentów należy wykonać badanie genotypu CYP2C9, aby określić aktywność izoenzymu CYP2C9 (patrz punkt 4.2). Pacjenci o homozygotycznym genotypie CYP2C9*3 (genotyp CYP2C9*3*3: około 0,3 do 0,4% populacji) nie powinni być leczeni siponimodem. Stosowanie siponimodu u tych pacjentów powoduje znaczne zwiększenie stężenia siponimodu w osoczu. Aby uniknąć zwiększonej ekspozycji na siponimod zalecana dawka podtrzymująca wynosi 1 mg na dobę u pacjentów z genotypem CYP2C9*2*3 (1,4-1,7% populacji) oraz u pacjentów z genotypem *1*3 (9-12% populacji) (patrz punkty 4.2 i 5.2).
Kobiety w wieku rozrodczym
Ze względu na ryzyko dla płodu siponimod jest przeciwwskazany podczas ciąży oraz u kobiet w wieku rozrodczym niestosujących skutecznej antykoncepcji. Przed rozpoczęciem leczenia kobiety w wieku rozrodczym muszą zostać poinformowane o tym ryzyku dla płodu, muszą uzyskać negatywny wynik testu ciążowego i stosować skuteczną antykoncepcję podczas leczenia i przez co najmniej 10 dni po zakończeniu leczenia (patrz punkty 4.3 i 4.6).
Przerwanie leczenia siponimodem
Po przerwaniu leczenia innym modulatorem receptora S1P rzadko zgłaszano występowanie ciężkiego zaostrzenia choroby, w tym efektu z odbicia. Należy wziąć pod uwagę możliwość wystąpienia ciężkiego zaostrzenia choroby po przerwaniu leczenia siponimodem. Należy obserwować pacjentów, czy nie występują u nich istotne objawy możliwego ciężkiego zaostrzenia choroby lub nawrotu dużej aktywności choroby po zakończeniu leczenia siponimodem, i w razie konieczności wdrożyć odpowiednie leczenie.
Po przerwaniu leczenia siponimodem lek utrzymuje się we krwi przez okres do 10 dni. Rozpoczęcie innego leczenia w tym czasie spowoduje jednoczesną ekspozycję na siponimod.
Po zakończeniu leczenia siponimodem z powodu PML zaleca się monitorowanie pacjentów pod kątem wystąpienia zapalnego zespołu rekonstrukcji immunologicznej (PML-IRIS) (patrz punkt „Postępująca wieloogniskowa leukoencefalopatia” powyżej).
U zdecydowanej większości (90%) pacjentów z SPMS liczba limfocytów powraca do zakresu wartości prawidłowych w ciągu 10 dni od zakończenia leczenia. Jednak resztkowe działania farmakodynamiczne, takie jak zmniejszenie liczby limfocytów obwodowych, mogą utrzymywać się do 3-4 tygodni po przyjęciu ostatniej dawki. Stosowanie leków immunosupresyjnych w tym okresie może spowodować działania addycyjne na układ immunologiczny i dlatego należy zachować ostrożność przez 3 do 4 tygodni po przyjęciu ostatniej dawki.
Wpływ na wyniki badań hematologicznych
Siponimod zmniejsza liczbę limfocytów we krwi poprzez ich redystrybucję do wtórnych narządów limfatycznych, dlatego liczby limfocytów we krwi obwodowej nie można wykorzystywać do oceny subpopulacji limfocytów u pacjentów leczonych siponimodem. Badania laboratoryjne z wykorzystaniem krążących komórek jednojądrzastych wymagają pobrania większej objętości krwi ze względu na zmniejszenie liczby krążących limfocytów.
Substancje pomocnicze
Tabletki zawierają lecytynę sojową. Pacjenci z nadwrażliwością na orzeszki ziemne lub soję nie powinni przyjmować siponimodu (patrz punkt 4.3).
Tabletki zawierają laktozę. Pacjenci z rzadko wystęującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy nie powinni przyjmować tego produktu leczniczego.
Leki przeciwnowotworowe, immunomodulujące lub immunosupresyjne
Nie badano działania siponimodu w skojarzeniu z lekami przeciwnowotworowymi, immunomodulującymi lub immunosupresyjnymi. Należy zachować ostrożność podczas jednoczesnego stosowania tych leków ze względu na ryzyko ich addycyjnego działania na układ immunologiczny, a także w tygodniach po zakończeniu podawania tych produktów leczniczych (patrz punkt 4.4). Ze względu na rodzaj i czas trwania immunosupresyjnych działań alemtuzumabu opisanych w informacji o tym produkcie, nie zaleca się rozpoczynania leczenia siponimodem po leczeniu alemtuzumabem, chyba że korzyści z leczenia wyraźnie przewyższają ryzyko dla danego pacjenta (patrz punkt 4.4).
Przeciwarytmiczne produkty lecznicze, produkty lecznicze wydłużające odstęp QT, produkty lecznicze mogące zmniejszać częstość akcji serca
Podczas rozpoczynania leczenia siponimodu nie należy stosować u pacjentów przyjmujących jednocześnie przeciwarytmiczne produkty lecznicze klasy Ia (np. chinidynę, prokainamid) lub klasy III (np. amiodaron, sotalol), produkty lecznicze wydłużające odstęp QT o znanych właściwościach arytmogennych, leki blokujące kanały wapniowe zmniejszające częstość akcji serca (takie jak werapamil lub diltiazem) lub inne substancje mogące zmniejszać częstość akcji serca (np. iwabradynę lub digoksynę) z powodu potencjalnego addycyjnego wpływu na częstość akcji serca (patrz punkt 4.4). Brak dostępnych danych dotyczących jednoczesnego stosowania tych produktów leczniczych z siponimodem. Jednoczesne stosowanie tych substancji podczas rozpoczynania leczenia może być związane z wystąpieniem ciężkiej bradykardii i bloku serca. Ze względu na potencjalny addytywny wpływ na częstość akcji serca leczenia siponimodem na ogół nie należy rozpoczynać u pacjentów leczonych jednocześnie tymi substancjami (patrz punkt 4.4). Jeśli rozważa się leczenie siponimodem, należy skonsultować się z kardiologiem w kwestii zmiany leczenia na produkty lecznicze, które nie zmniejszają częstości akcji serca, lub w kwestii odpowiedniego monitorowania stanu pacjenta podczas rozpoczynania leczenia.
Leki beta-adrenolityczne
Należy zachować ostrożność, gdy leczenie siponimodem jest rozpoczynane u pacjentów otrzymujących leki beta-adrenolityczne ze względu na addycyjne działanie zmniejszające częstość akcji serca (patrz punkt 4.4). Leczenie lekami beta-adrenolitycznymi może być rozpoczynane u pacjentów otrzymujących stałe dawki siponimodu.
Ujemne działanie chronotropowe spowodowane jednoczesnym podawaniem siponimodu i propranololu oceniano w specjalnym badaniu farmakodynamicznym/bezpieczeństwa stosowania. Dołączenie propranololu po osiągnięciu przez siponimod stanu stacjonarnego w odniesieniu do parametrów farmakodynamicznych/farmakokinetycznych powodowało mniejsze ujemne działanie chronotropowe (mniej niż addycyjne) niż w sytuacji, gdy siponimod dołączano do schematu leczenia po osiągnięciu stanu stacjonarnego przez propranolol w odniesieniu do jego parametrów farmakokinetycznych/farmakodynamicznych (addycyjny wpływ na częstość akcji serca).
Szczepienia
Stosowanie żywych szczepionek atenuowanych może nieść ze sobą ryzyko wystąpienia zakażeń i dlatego należy unikać tych szczepionek podczas leczenia siponimodem i przez okres 4 tygodni po leczeniu (patrz punkt 4.4).
Podczas leczenia siponimodem i przez okres do 4 tygodni po leczeniu szczepienia mogą być mniej skuteczne. Uważa się, że skuteczność szczepienia nie zmniejszy się, jeśli leczenie siponimodem zostanie przerwane na 1 tydzień przed szczepieniem aż do upływu 4 tygodni po szczepieniu. W dedykowanym badaniu fazy I u zdrowych ochotników, jednoczesne leczenie siponimodem i podawanie szczepionek przeciw grypie lub krótsza przerwa w leczeniu (od 10 dni do nie więcej niż 14 dni po szczepieniu) wykazały gorszy odsetek odpowiedzi (około 15% do 30% mniejszy) w porównaniu z placebo, podczas gdy skuteczność szczepienia PPV 23 nie była zmieniona przez jednoczesne leczenie siponimodem (patrz punkt 4.4).
Potencjalny wpływ innych produktów leczniczych na farmakokinetykę siponimodu
Siponimod jest metabolizowany głównie przez cytochrom P450 2C9 (CYP2C9) (79,3%), a w mniejszym stopniu przez cytochrom P450 3A4 (CYP3A4) (18,5%). CYP2C9 jest enzymem polimorficznym i przewiduje się, że wpływ interakcji między lekami w obecności leków będących inhibitorami lub induktorami CYP3A lub CYP2C9 będzie zależeć od genotypu CYP2C9.
Inhibitory CYP2C9 i CYP3A4
Ze względu na istotne zwiększenie ekspozycji na siponimod, nie zaleca się jednoczesnego stosowania siponimodu i produktów leczniczych będących umiarkowanymi inhibitorami CYP2C9 oraz umiarkowanymi lub silnymi inhibitorami CYP3A4. Ten schemat jednoczesnego podawania może obejmować umiarkowany podwójny inhibitor CYP2C9/CYP3A4 (np. flukonazol) lub umiarkowany inhibitor CYP2C9 w skojarzeniu z oddzielnym umiarkowanym lub silnym inhibitorem CYP3A4.
Jednoczesne podawanie flukonazolu (umiarkowanego inhibitora CYP2C9/podwójnego inhibitora CYP3A4) w dawce 200 mg na dobę w stanie stacjonarnym oraz pojedynczej dawki 4 mg siponimodu zdrowym ochotnikom z genotypem CYP2C9*1*1 spowodowało 2-krotne zwiększenie pola powierzchni pod krzywą (AUC) siponimodu. Zgodnie z oceną potencjalnych interakcji leków za pomocą modelowania farmakokinetycznego opartego na danych fizjologicznych (ang. physiologically based pharmacokinetic, PBPK), przewiduje się wystąpienie maksymalnie 2-krotnego zwiększenia AUC siponimodu we wszystkich genotypach po podaniu dowolnego rodzaju inhibitora CYP3A4 i CYP2C9, z wyjątkiem pacjentów z genotypem CYP2C9*2*2. U pacjentów z genotypem CYP2C9*2*2 przewiduje się 2,7-krotne zwiększenie AUC siponimodu w obecności umiarkowanych inhibitorów CYP2C9/CYP3A4.
Induktory CYP2C9 i CYP3A4
Siponimod można podawać w skojarzeniu z większością rodzajów induktorów CYP2C9 i CYP3A4. Jednak ze względu na spodziewane zmniejszenie ekspozycji na siponimod, należy rozważyć stosowność i możliwe korzyści z leczenia, gdy siponimod jest podawany w skojarzeniu:
W tych warunkach przewiduje się istotne zmniejszenie ekspozycji na siponimod (odpowiednio o 76% i 51%), na podstawie oceny potencjalnych interakcji między lekami z użyciem modelowania PBPK. Jednoczesne podawanie siponimodu w dawce 2 mg na dobę w obecności dobowych dawek 600 mg ryfampicyny (silnego induktora CYP3A4 i umiarkowanego induktora CYP2C9) zmniejszało AUCtau,ss i Cmax,ss siponimodu odpowiednio o 57% i 45% u pacjentów z genotypem CYP2C9*1*1.
Doustne środki antykoncepcyjne
Jednoczesne podawanie siponimodu nie ujawniło klinicznie istotnego wpływu na farmakokinetykę i farmakodynamikę złożonego doustnego środka antykoncepcyjnego zawierającego etynyloestradiol i lewonorgestrel. Z tego względu skuteczność badanego doustnego środka antykoncepcyjnego była zachowana podczas leczenia siponimodem.
Nie przeprowadzono badań dotyczących interakcji z doustnymi środkami antykoncepcyjnymi zawierającymi inne progestageny, jednak nie oczekuje się, aby siponimod miał wpływ na skuteczność doustnych środków antykoncepcyjnych.
Kobiety w wieku rozrodczym / Antykoncepcja u kobiet
Siponimod jest przeciwwskazany u kobiet w wieku rozrodczym niestosujących skutecznej antykoncepcji (patrz punkt 4.3). Dlatego przed rozpoczęciem leczenia u kobiet w wieku rozrodczym należy uzyskać ujemny wynik testu ciążowego oraz zapewnić poradnictwo dotyczące poważnego ryzyka dla płodu. Kobiety w wieku rozrodczym muszą stosować skuteczną antykoncepcję podczas leczenia i przez co najmniej dziesięć dni po przyjęciu ostatniej dawki siponimodu (patrz punkt 4.4).
Specjalne sposoby postępowania przedstawione są również w pakiecie edukacyjnym dla lekarzy. Te procedury należy wdrożyć, zanim siponimod zostanie przepisany pacjentkom i podczas leczenia.
Przerywając leczenie siponimodem z powodu planowanej ciąży należy wziąć pod uwagę możliwy nawrót aktywności choroby (patrz punkt 4.4).
Ciąża
Brak danych lub istnieją ograniczone dane dotyczące stosowania siponimodu u kobiet w ciąży. Badania na zwierzętach wykazały toksyczne działania na zarodek i płód szczurów i królików, wywołane przez siponimod oraz działania teratogenne u szczurów, w tym obumarcie zarodka i płodu oraz wady rozwojowe kośćca lub narządów trzewnych przy ekspozycji porównywalnej do ekspozycji występującej u ludzi po podaniu dawki dobowej wynoszącej 2 mg (patrz punkt 5.3). Ponadto doświadczenie kliniczne z innym modulatorem receptora sfingozyno-1-fosforanu wskazywało na 2- krotnie większe ryzyko ciężkich wad wrodzonych, gdy lek był podawany podczas ciąży w porównaniu z częstością występowania tych wad w populacji ogólnej.
W związku z tym siponimod jest przeciwwskazany podczas ciąży (patrz punkt 4.3). Podawanie siponimodu należy przerwać co najmniej 10 dni przed planowanym zajściem w ciążę (patrz punkt 4.4). Jeśli kobieta zajdzie w ciążę podczas leczenia, siponimod należy odstawić. Należy udzielić pacjentce porady medycznej dotyczącej ryzyka szkodliwego wpływu na płód w związku z leczeniem i wykonać badania ultrasonograficzne.
Karmienie piersią
Nie wiadomo, czy siponimod lub jego główne metabolity przenikają do mleka kobiecego. Siponimod i jego metabolity przenikają do mleka szczurów. Siponimodu nie należy stosować podczas karmienia piersią.
Płodność
Nie oceniano wpływu siponimodu na płodność ludzi. Siponimod nie ma wpływu na narządy rozrodcze samców szczura i małpy ani na parametry płodności u szczurów.
Siponimod nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednak sporadycznie mogą wystąpić zawroty głowy podczas rozpoczynania leczenia siponimodem. Dlatego pacjenci nie powinni prowadzić pojazdów ani obsługiwać maszyn w pierwszym dniu po rozpoczęciu leczenia siponimodem (patrz punkt 4.4).
Podsumowanie profilu bezpieczeństwa
Profil bezpieczeństwa siponimodu scharakteryzowano na podstawie danych pochodzących z głównego badania klinicznego. Najczęstszymi działaniami niepożądanymi stwierdzonymi w głównej części badania A2304 były: ból głowy (15%) i nadciśnienie tętnicze (12,6%). Informacje dotyczące bezpieczeństwa pochodzące z przedłużenia długoterminowego badania A2304 były zgodne z obserwowanymi w części głównej badania.
Tabelaryczny wykaz działań niepożądanych
W obrębie każdej grupy układów i narządów działania niepożądane przedstawiono według częstości występowania, zaczynając od najczęściej występujących. Ponadto, częstość występowania każdego działania niepożądanego opiera się na następującej konwencji: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1 000 do <1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000); nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 2 Tabelaryczny wykaz działań niepożądanych
|
Zakażenia i zarażenia pasożytnicze |
|
|
Często |
Półpasiec |
|
Nieznana |
Kryptokokowe zapalenie opon mózgowych Postępująca wieloogniskowa leukoencefalopatia |
|
Nowotwory łagodne, złośliwe i nieokreślone (w tym torbiele i polipy) |
|
|
Często |
Znamiona barwnikowe Rak podstawnokomórkowy |
|
Niezbyt często |
Rak kolczystokomórkowy Czerniak złośliwy |
|
Zaburzenia krwi i układu chłonnego |
|
|
Często |
Limfopenia |
| Zaburzenia układu immunologicznego | |
| Rzadko | Zapalny zespół rekonstrukcji immunologicznej (IRIS) |
|
Zaburzenia układu nerwowego |
|
|
Bardzo często |
Ból głowy |
|
Często |
Zawroty głowy Napady drgawkowe Drżenie |
|
Zaburzenia oka |
|
|
Często |
Obrzęk plamki żółtej |
|
Zaburzenia serca |
|
|
Często |
Bradykardia Blok przedsionkowo-komorowy (pierwszego i drugiego stopnia) |
|
Zaburzenia naczyniowe |
|
|
Bardzo często |
Nadciśnienie tętnicze |
|
Zaburzenia żołądka i jelit |
|
|
Często |
Nudności Biegunka |
|
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej |
|
|
Często |
Ból kończyny |
|
Zaburzenia ogólne i stany w miejscu podania |
|
|
Często |
Obrzęk obwodowy Astenia |
|
Badania diagnostyczne |
|
|
Bardzo często |
Zwiększenie wyników badań czynności wątroby |
|
Często |
Zmniejszenie wyników badań czynności płuc |
Opis wybranych działań niepożądanych
Zakażenia
W badaniu klinicznym III fazy z udziałem pacjentów z SPMS całkowita częstość występowania zakażeń była porównywalna u pacjentów leczonych siponimodem i u pacjentów otrzymujących placebo (odpowiednio 49,0% w porównaniu z 49,1%). Jednak zgłoszono zwiększenie częstości występowania zakażeń wirusem półpaśca u pacjentów leczonych siponimodem (2,5%) w porównaniu z placebo (0,7%).
Po zastosowaniu siponimodu w dowolnym momencie podczas leczenia występowały przypadki zapalenia opon mózgowych lub zapalenia opon mózgowych i mózgu spowodowane przez wirusy ospy wietrznej i półpaśca. Po zastosowaniu siponimodu zgłaszano także przypadki kryptokokowego zapalenia opon mózgowych (ang. cryptococcal meningitis, CM) (patrz punkt 4.4).
Obrzęk plamki żółtej
Obrzęk plamki żółtej był częściej zgłaszany u pacjentów otrzymujących siponimod (1,8%) niż u pacjentów otrzymujących placebo (0,2%). Chociaż większość przypadków wystąpiła w ciągu 3 do 4 miesięcy od rozpoczęcia leczenia siponimodem, zgłaszano także przypadki występujące u pacjentów leczonych siponimodem przez ponad 6 miesięcy (patrz punkt 4.4). Niektórzy pacjenci zgłaszali takie objawy jak nieostre widzenie lub pogorszenie ostrości wzroku, natomiast u innych pacjentów obrzęk plamki żółtej był bezobjawowy i został rozpoznany podczas rutynowego badania okulistycznego. Obrzęk plamki żółtej na ogół zmniejszał się lub ustępował samoistnie po przerwaniu leczenia. Nie oceniano ryzyka nawrotu po wznowieniu leczenia.
Bradyarytmia
Rozpoczęcie leczenia siponimodem powoduje przemijające spowolnienie częstości akcji serca i może być również związane ze zwolnieniem przewodzenia przedsionkowo-komorowego (patrz punkt 4.4). Bradykardię zgłaszano u 6,2% pacjentów leczonych siponimodem w porównaniu z 3,1% pacjentów otrzymujących placebo, a blok AV zgłaszano u 1,7% pacjentów leczonych siponimodem w porównaniu z 0,7% pacjentów otrzymujących placebo (patrz punkt 4.4).
Maksymalne zmniejszenie częstości akcji serca obserwuje się w ciągu pierwszych 6 godzin po podaniu dawki leku.
Przemijające, zależne od dawki zmniejszenie częstości akcji serca obserwowano w początkowej fazie podawania leku, a efekt plateau występował po podaniu dawek ≥5 mg. Zdarzenia bradyarytmii (bloki AV i zahamowania zatokowe) były wykrywane częściej podczas leczenia siponimodem w porównaniu z placebo.
Większość bloków AV i zahamowań zatokowych występowało po podaniu dawek większych od dawki terapeutycznej wynoszącej 2 mg, ze znacznie większą częstością występowania tych zdarzeń u pacjentów niepoddanych stopniowemu zwiększaniu dawki niż u pacjentów stopniowo zwiększających dawkę do dawki podtrzymującej.
Zmniejszenie częstości akcji serca wywołane siponimodem można odwrócić za pomocą atropiny lub izoprenaliny.
Badania czynności wątroby
U pacjentów z SM leczonych siponimodem zgłaszano zwiększenie aktywności enzymów wątrobowych (głównie zwiększenie aktywności AlAT). W badaniu III fazy u pacjentów z SPMS podwyższone wyniki prób wątrobowych były obserwowane częściej u pacjentów leczonych siponimodem (11,3%) niż u pacjentów otrzymujących placebo (3,1%), głównie z powodu zwiększonej aktywności aminotransferaz wątrobowych (AlAT/AspAT) i GGT. W większości przypadków zwiększona aktywność wystąpiła w ciągu 6 miesięcy od rozpoczęcia leczenia. Aktywność AlAT powróciła do normy w ciągu około 1 miesiąca po przerwaniu leczenia siponimodem (patrz punkt 4.4).
Ciśnienie krwi
W badaniu klinicznym III fazy z udziałem pacjentów z SPMS nadciśnienie tętnicze zgłaszano częściej u pacjentów leczonych siponimodem (12,6%) niż u pacjentów otrzymujących placebo (9,0%). Leczenie siponimodem powodowało zwiększenie skurczowego i rozkurczowego ciśnienia krwi występujące na wczesnym etapie po rozpoczęciu leczenia. Działanie to osiągnęło maksymalne nasilenie po około 6 miesiącach leczenia (ciśnienie skurczowe 3 mmHg, ciśnienie rozkurczowe 1,2 mmHg) i pozostawało stabilne w późniejszym okresie. Działanie to utrzymywało się w miarę kontynuowania leczenia.
Napady drgawkowe
Napady drgawkowe zgłaszano u 1,7% pacjentów leczonych siponimodem w porównaniu z 0,4% pacjentów otrzymujących placebo w badaniu klinicznym III fazy z udziałem pacjentów z SPMS.
Działania na układ oddechowy
Podczas leczenia siponimodem obserwowano nieznaczne zmniejszenie natężonej objętości wydechowej pierwszosekundowej (FEV1 – ang. forced expiratory volume in 1 second) i zdolności dyfuzyjnej płuc dla tlenku węgla (ang. DLCO – diffusing capacity of the lung for carbon monoxide). W badaniu klinicznym III fazy z udziałem pacjentów z SPMS w 3. i 6. miesiącu leczenia średnie zmiany w FEV1 w stosunku do wartości wyjściowej w grupie leczonej siponimodem wyniosły -0,1 l w każdym punkcie czasowym, przy braku zmian w grupie placebo. Te obserwacje wskazywały na nieco większe wartości (średnia zmiana w FEV1 o około 0,15 l w stosunku do wartości wyjściowej) u pacjentów z zaburzeniami układu oddechowego, takimi jak przewlekła obturacyjna choroba płuc (POChP) lub astma, leczonych siponimodem. Podczas długotrwałego leczenia to zmniejszenie nie przekładało się na klinicznie istotne zdarzenia niepożądane i nie wiązało się ze zwiększeniem liczby zgłaszanych przypadków kaszlu lub duszności (patrz punkt 5.1).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
U osób zdrowych pojedynczą maksymalną dawkę tolerowaną określono na 25 mg na podstawie występowania objawowej bradykardii po podaniu pojedynczych dawek 75 mg. Kilka osób otrzymało niezamierzone dawki do 200 mg na dobę przez 3 do 4 dni i wystąpiło u nich bezobjawowe, łagodne do umiarkowanego zwiększenie wyników badań czynności wątroby.
U jednego pacjenta (z depresją w wywiadzie), który przyjął 84 mg siponimodu wystąpiło niewielkie zwiększenie aktywności aminotransferaz wątrobowych.
Jeśli przedawkowanie jest jednocześnie pierwszym narażeniem na siponimod lub występuje w fazie stopniowego zwiększania dawki siponimodu, ważne jest, aby monitorować stan pacjenta pod kątem przedmiotowych i podmiotowych objawów bradykardii, co może obejmować monitorowanie pacjenta przedłużone do następnego dnia. Konieczne są regularne pomiary tętna i ciśnienia krwi oraz wykonanie badania elektrokardiograficznego (patrz punkty 4.2 i 4.4).
Nie ma specyficznego antidotum dla siponimodu. Siponimod nie jest w znaczącym stopniu usuwany z organizmu ani poprzez dializę, ani poprzez wymianę osocza
Grupa farmakoterapeutyczna: Leki immunosupresyjne, modulatory receptora sfingozyno-1-fosforanu (S1P), kod ATC: L04AE03
Mechanizm działania
Siponimod jest modulatorem receptora sfingozyno-1-fosforanu (S1P). Siponimod wiąże się wybiórczo z dwoma z pięciu receptorów sprzężonych z białkami G (GPCR) dla S1P, czyli S1P1 i S1P5. Działając jako czynnościowy antagonista receptorów S1P1 na limfocytach, siponimod zapobiega wyjściu limfocytów z węzłów chłonnych. Takie działanie zmniejsza ponowne krążenie limfocytów T w ośrodkowym układzie nerwowym (OUN), aby ograniczyć tam stan zapalny.
Działanie farmakodynamiczne
Zmniejszenie liczby limfocytów we krwi obwodowej
Siponimod wywołuje zależne od dawki zmniejszenie liczby limfocytów we krwi obwodowej w ciągu 6 godzin od podania pierwszej dawki, spowodowane odwracalnym zatrzymaniem limfocytów w tkankach limfatycznych.
Podczas ciągłego codziennego stosowania liczba limfocytów stale zmniejsza się osiągając najmniejszą wartość o medianie (90% CI) liczby limfocytów wynoszącej około 0,560 (0,271-1,08) komórek/nl u typowego pacjenta z SPMS niebędącego pochodzenia japońskiego i z genotypem CYP2C9*1*1 lub *1*2, co odpowiada 20-30% wartości wyjściowej. Mała liczba limfocytów utrzymuje się podczas codziennego przyjmowania leku.
U zdecydowanej większości (90%) pacjentów z SPMS liczba limfocytów powraca do zakresu wartości prawidłowych w ciągu 10 dni od zakończenia leczenia. Po zakończeniu leczenia siponimodem pozostałe (resztkowe) działanie polegające na zmniejszeniu liczby limfocytów we krwi obwodowej może utrzymywać się przez okres do 3-4 tygodni po przyjęciu ostatniej dawki.
Częstość akcji serca i rytm serca
Siponimod powoduje przemijające zmniejszenie częstości akcji serca i zwolnienie przewodzenia przedsionkowo-komorowego w chwili rozpoczynania leczenia (patrz punkt 4.4 i 4.8), które jest mechanistycznie związane z aktywacją dokomórkowych prostowniczych kanałów potasowych sprzężonych z białkiem G (ang. G-protein-coupled inwardly rectifying potassium, GIRK) poprzez stymulację receptora S1P1 prowadzącą do hiperpolaryzacji komórek i zmniejszonej pobudliwości. Ze względu na swój czynnościowy antagonizm w receptorach S1P1, początkowe stopniowe zwiększanie dawki siponimodu sukcesywnie zmniejsza czułość kanałów GIRK aż do osiągnięcia dawki podtrzymującej.
Możliwość wydłużenia odstępu QT
W dokładnym badaniu odstępu QT oceniano wpływ terapeutycznych (2 mg) i supraterapeutycznych (10 mg) dawek siponimodu na repolaryzacje serca. Wyniki nie sugerowały działania arytmogennego związanego z wydłużeniem odstępu QT po zastosowaniu siponimodu. Siponimod zwiększał odstęp QTcF skorygowany względem placebo i dostosowany z uwzględnieniem wartości wyjściowych (ΔΔQTcF) o ponad 5 ms, przy czym maksymalny średni efekt wynosił odpowiednio 7,8 ms (2 mg) i 7,2 ms (10 mg) 3 godziny po podaniu dawki leku. Górna granica jednostronnego 95% CI dla ΔΔQTcF we wszystkich punktach czasowych pozostała poniżej 10 ms. Analiza kategorialna wykazała brak wartości QTc powyżej 480 ms w trakcie leczenia, brak zwiększenia QTc o ponad 60 ms względem wartości wyjściowych oraz brak skorygowanych lub nieskorygowanych wartości QT/QTc większych niż 500 ms.
Czynność płuc
Leczenie siponimodem w dawkach pojedynczych lub wielokrotnych podawanych przez 28 dni nie jest związane z klinicznie istotnym zwiększeniem oporu dróg oddechowych mierzonym za pomocą natężonej objętości wydechowej pierwszosekundowej (FEV1) i natężonego przepływu wydechowego (FEF) podczas wydychania 25 do 75% natężonej pojemności życiowej (FEF25-75%). Obserwowano nieznaczną tendencję do zmniejszania się FEV1 po podaniu pojedynczych dawek nieterapeutycznych (>10 mg). Wielokrotne dawki siponimodu były związane z łagodnymi do umiarkowanych zmianami FEV1 i FEF25-75%, które nie były zależne od dawki ani pory dnia i nie były związane z występowaniem jakichkolwiek klinicznych objawów zwiększonego oporu dróg oddechowych.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność siponimodu była analizowana w badaniu III fazy oceniającym dawki 2 mg podawane raz na dobę pacjentom z SPMS.
Badanie A2304 (EXPAND) w SPMS
Część główna badania A2304 była randomizowanym badaniem III fazy kontrolowanym placebo, prowadzonym metodą podwójnie ślepej próby, zależnym od zdarzeń i czasu trwania obserwacji, w którym uczestniczyli pacjenci z SPMS i udokumentowaną progresją choroby w ciągu ostatnich 2 lat przy braku nawrotów lub niezależnie od nawrotów, braku dowodów potwierdzających nawrót choroby w ciągu 3 miesięcy poprzedzających włączenie do badania oraz z wynikiem od 3,0 do 6,5 w rozszerzonej skali niewydolności ruchowej (ang. Expanded Disability Status Scale, EDSS) w chwili przystąpienia do badania. Mediana wyniku EDSS wyniosła 6,0 przed rozpoczęciem leczenia w badaniu. Pacjenci w wieku powyżej 61 lat nie byli włączani do badania. W odniesieniu do aktywności choroby, cechy charakterystyczne dla aktywności zapalnej w SPMS mogą być związane z nawrotem lub wynikiem badania obrazowego (tj. zmiany w obrazach T1-zależnych ulegające wzmocnieniu po podaniu gadolinu lub aktywne [nowe bądź powiększone] zmiany w obrazach T2-zależnych).
Pacjenci byli losowo przydzielani w stosunku 2:1 do grupy otrzymującej siponimod w dawce 2 mg raz na dobę lub placebo. Oceny kliniczne przeprowadzano podczas badań przesiewowych i co 3 miesiące oraz w momencie nawrotu choroby. Oceny MRI wykonywano podczas badań przesiewowych i co 12 miesięcy.
Pierwszorzędowym punktem końcowym badania był czas do 3-miesięcznej potwierdzonej progresji niesprawności (ang. confirmed disability progression, CDP) definiowanej jako zwiększenie o co najmniej 1 punkt wyniku w EDSS w stosunku do wartości wyjściowej (zwiększenie o 0,5 punktu w przypadku pacjentów z wyjściowym wynikiem w EDSS = 5,5 lub wyższym) utrzymujące się przez 3 miesiące. Najważniejszymi drugorzędowymi punktami końcowymi były: czas do 3-miesięcznego potwierdzonego pogorszenia o co najmniej 20% w teście szybkości chodu na odcinku 7,5 m (ang. timed 25-foot walk test, T25W) w stosunku do stanu wyjściowego oraz zmiana w objętości zmian w obrazach T2-zależnych względem wartości wyjściowej. Dodatkowe drugorzędowe punkty końcowe obejmowały czas do 6-miesięcznej CDP, procentową zmianę w objętości mózgu oraz miary aktywności zapalnej choroby (roczny wskaźnik rzutów, zmiany w badaniu MRI). Eksploracyjnym punktem końcowym była zmiana w prędkości przetwarzania będąca elementem procesu poznawczego, mierzona testem przyporządkowania symboli cyfrom (ang. Symbol Digit Modality Test).
Czas trwania badania był różny dla poszczególnych pacjentów (mediana czasu trwania badania wyniosła 21 miesięcy, zakres: od 1 dnia do 37 miesięcy).
W badaniu 1 651 pacjentów zostało losowo przydzielonych do leczenia siponimodem w dawce 2 mg (N=1 105) lub placebo (N=546); badanie ukończyło 82% pacjentów leczonych siponimodem i 78% pacjentów otrzymujących placebo. Mediana wieku wyniosła 49 lat, mediana czasu trwania choroby wyniosła 16 lat, a mediana wyniku w EDSS wyniosła 6,0 przed rozpoczęciem leczenia w badaniu. U 64% pacjentów nie występowały nawroty choroby w ciągu 2 lat przed przystąpieniem do badania, a u 76% pacjentów nie występowały zmiany ulegające wzmocnieniu po podaniu gadolinu (Gd) w wyjściowym badaniu MRI. 78% pacjentów otrzymywało wcześniej leczenie z powodu SM.
Czas do początku 3-miesięcznej i 6-miesięcznej CDP był istotnie opóźniony w przypadku leczenia siponimodem, przy zmniejszeniu ryzyka 3-miesięcznej CDP o 21% w porównaniu z placebo (hazard względny [HR] 0,79, p=0,0134) i zmniejszeniu ryzyka 6-miesięcznej CDP o 26% w porównaniu z placebo (HR 0,74, p=0,0058).
Rycina 1 Pacjenci z 3- i 6-miesięczną CDP na podstawie krzywych EDSS-Kaplana-Meiera (pełna analizowana grupa, badanie A2304)
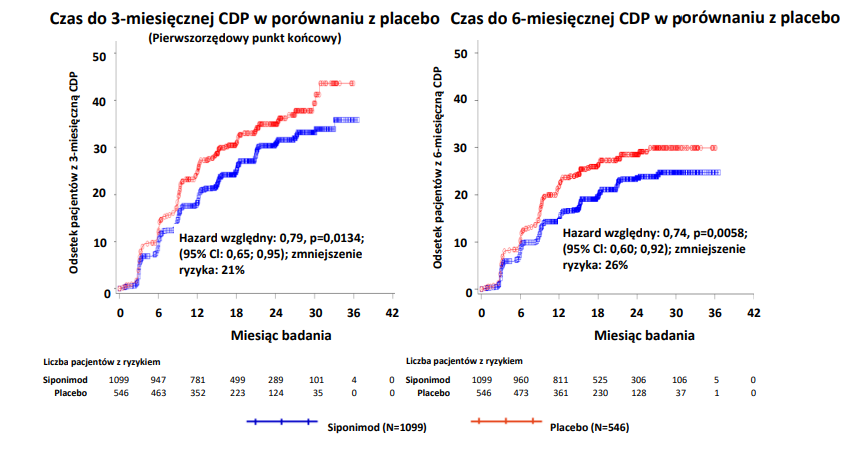
Tabela 3 Wyniki kliniczne i wyniki obrazowania MRI w badaniu A2304
|
Punkty końcowe |
A2304 (EXPAND) |
|
|
Siponimod 2 mg |
Placebo |
|
|
Kliniczne punkty końcowe |
||
|
Pierwszorzędowy punkt końcowy oceny skuteczności: |
26,3% |
31,7% |
|
Zmniejszenie ryzyka1 |
21% (p=0,0134) |
|
|
Odsetek pacjentów z 3-miesięcznym potwierdzonym 20% wydłużeniem testu szybkości chodu na odcinku 7,5 m |
39,7% |
41,4% |
|
Zmniejszenie ryzyka1 |
6% (p=0,4398) |
|
|
Odsetek pacjentów z 6-miesięczną potwierdzoną progresją niesprawności |
19,9% |
25,5% |
|
Zmniejszenie ryzyka1 |
26% [(p=0,0058)]6 |
|
|
Roczny wskaźnik rzutów (ang. annualised relapse rate, ARR) |
0,071 |
0,152 |
|
Zmniejszenie wskaźnika2 |
55% [(p<0,0001)] 6><0,0001)]6 |
|
|
Punkty końcowe w badaniu MRI |
||
|
Zmiana w objętości zmian w obrazach T2-zależnych względem wartości wyjściowych (mm3 ) 3 |
+184 mm3 |
+879 mm3 |
|
Różnica w zmianie dotyczącej objętości zmian w obrazach T2-zależnych |
-695 mm3 (p<0,0001) 7><0,0001)7 |
|
|
Odsetkowa zmiana objętości mózgu względem stanu wyjściowego (95% CI)3 |
-0,497% |
-0,649% |
|
Różnica w odsetkowej zmianie objętości mózgu |
0,152% [(p=0,0002)] 6 |
|
|
Średnia skumulowana liczba zmian w obrazach T1-zależnych, ulegających wzmocnieniu po podaniu Gd (95% CI)4 |
0,081 |
0,596 |
|
Zmniejszenie odsetka |
86% [(p<0,0001)]6 |
|
|
Odsetek pacjentów z 4-punktowym pogorszeniem wyniku testu przyporządkowania symboli cyfrom5 |
16,0% |
20,9% |
|
Zmniejszenie ryzyka1 |
25% [(p=0,0163)]6 |
|
|
1 Na podstawie modelowania Coxa dotyczącego czasu do progresji 2 Na podstawie modelu dla zdarzeń nawracających 3 Średnia z miesiąca 12. i miesiąca 24. 4 Do miesiąca 24. 5 Potwierdzony po 6 miesiącach 6 [Nominalna wartość p dla punktów końcowych nie została uwzględniona w procedurze hierarchicznego testowania i nie została skorygowana z powodu wielokrotności porównań] 7 Niekonfirmacyjna wartość p; procedura hierarchicznego testowania zakończona przed osiągnięciem punktu końcowego |
||
Wyniki badania wykazały zmienne, ale konsekwentne zmniejszenie ryzyka dotyczące czasu do 3- i 6-miesięcznej CDP po zastosowaniu siponimodu w porównaniu z placebo w podgrupach wyodrębnionych ze względu na płeć, wiek, aktywność nawrotów choroby przed badaniem, wyjściową aktywność choroby w badaniu MRI, czas trwania choroby i wyjściowy poziom niesprawności.
W podgrupie pacjentów (n=779) z chorobą aktywną (definiowanej jako pacjenci z nawrotem choroby w ciągu 2 lat poprzedzających badanie i (lub) wyjściową obecnością zmian w obrazach T1-zależnych ulegających wzmocnieniu po podaniu gadolin), charakterystyka wyjściowa była podobna, jak w całej populacji. Mediana wieku wyniosła 47 lat, mediana czasu trwania choroby wyniosła 15 lat, a mediana wyniku w EDSS na początku badania wyniosła 6,0.
Czas do początku 3-miesięcznej i 6-miesięcznej CDP był znamiennie opóźniony u pacjentów leczonych siponimodem z chorobą aktywną, odpowiednio o 31% w porównaniu z placebo (hazard względny [HR] 0,69; 95% CI: 0,53, 0,91) i o 37% w porównaniu z placebo (HR 0,63; 95% CI: 0,47, 0,86). ARR (nawroty potwierdzone) zmniejszył się o 46% (stosunek ARR 0,54; 95% CI: 0,39, 0,77) w porównaniu z placebo. Względne zmniejszenie odsetka skumulowanej liczby zmian w obrazach T1- zależnych ulegających wzmocnieniu po podaniu gadolinu w ciągu 24 miesięcy wyniosła 85% (stosunek odsetków 0,155; 95% CI: 0,104, 0,231) w porównaniu z placebo. Różnice w zmianie objętości zmian w obrazach T2-zależnych oraz w odsetku zmiany objętości mózgu (średnia z 12 i 24 miesięcy) w porównaniu z placebo wyniosły odpowiednio -1163 mm3 (95% CI: -1484, -843 mm3 ) oraz 0,141% (95% CI: 0,020, 0,261%).
Rycina 2 Pacjenci z 3- i 6-miesięczną CDP na podstawie krzywych Kaplana-Meiera dla EDSS - podgrupa z aktywną SPMS (pełna analizowana grupa, badanie A2304)
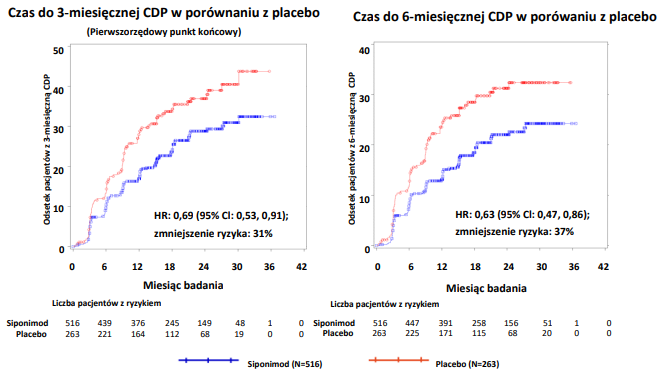
W podgrupie pacjentów (n=827) bez przedmiotowych i podmiotowych objawów aktywności choroby (definiowanej jako pacjenci bez nawrotu w ciągu 2 lat poprzedzających udział w badaniu i bez obecności wyjściowych zmian w obrazach T1-zależnych ulegających wzmocnieniu po podaniu Gd) wpływ na 3-miesięczną i 6-miesięczną CDP był mały (zmniejszenie ryzyka wyniosło odpowiednio 7% i 13%).
Analiza post-hoc badania A2304 (EXPAND) wykazała, że siponimod opóźnia progresję do EDSS ≥7,0 (utrzymując się do końca badania, tj. czasu przejścia na wózek inwalidzki), co skutkuje zmniejszeniem ryzyka o 38% (HR z modelu Coxa 0,62; 95% CI: 0,41, 0,92). Oszacowany metodą Kaplana-Meiera odsetek pacjentów, u których wystąpiła progresja do EDSS ≥7,0 w 24. miesiącu, wyniósł 6,97% w grupie leczonej siponimodem i 8,72% w grupie otrzymującej placebo. W podgrupie pacjentów z aktywnym SPMS zmniejszenie ryzyka wyniosło 51% (HR 0,49; 95% CI: 0,27; 0,90), a oszacowany metodą Kaplana-Meiera odsetek pacjentów w 24. miesiącu wyniósł 6,51% w grupie leczonej siponimodem i 8,69% w grupie otrzymującej placebo. Ponieważ wyniki te miały charakter eksploracyjny, należy je interpretować z ostrożnością.
Po części głównej badania A2304 przeprowadzono otwarte przedłużenie badania z jedną grupą terapeutyczną. Cel badania przedłużonego miał charakter eksploracyjny i polegał na ocenie długoterminowej skuteczności i bezpieczeństwa stosowania siponimodu, prowadzonej w trakcie leczenia trwającego dodatkowo do 7 lat. Z całej liczby pacjentów poddanych randomizacji, do przedłużenia badania włączono 68% (n=1 120) pacjentów, a 29% pacjentów (n=485) ukończyło przedłużenie badania A2304. Oszacowanie Kaplana-Meiera dotyczące odsetka pacjentów z 6-miesięczną CDP w miesiącu 108. wyniosło 64,7% w grupie stosującej nieprzerwanie siponimod i 68,4% w grupie pacjentów, którzy zmienili leczenie z placebo na siponimod po zakończeniu części głównej badania. U pacjentów z czynną postacią SPMS oszacowanie Kaplana-Meiera dotyczące odsetka pacjentów z 6-miesięczną CDP w miesiącu 108. wyniosło 62,9% w grupie nieprzerwanego leczenia siponimodem i 68,1% w grupie pacjentów, którzy zmienili leczenie z placebo na siponimod po zakończeniu części głównej badania.
Dzieci i młodzież
Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań siponimodu w jednej lub kilku podgrupach populacji dzieci i młodzieży w leczeniu stwardnienia rozsianego (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Wchłanianie
Czas (Tmax) do osiągnięcia maksymalnego stężenia w osoczu (Cmax) po wielokrotnym doustnym podaniu siponimodu wynosi około 4 godzin (zakres: 2 do 12 godzin). Siponimod jest wchłaniany w dużym stopniu (≥70%, na podstawie ilości radioaktywności wydalanej z moczem oraz ilości metabolitów w kale ekstrapolowanej do nieskończoności). Bezwzględna biodostępność siponimodu po podaniu doustnym wynosi około 84%. Po zastosowaniu siponimodu w dawce 2 mg podawanej raz na dobę przez 10 dni w dniu 10. średnie Cmax wyniosło 30,4 ng/ml, a średnie AUCtau wyniosło 558 h*ng/ml. Stan stacjonarny był osiągany po około 6 dniach wielokrotnego podawania siponimodu raz na dobę.
Pomimo opóźnienia w Tmax do 8 godzin po podaniu pojedynczej dawki spożycie pokarmu nie miało wpływu na ogólnoustrojową ekspozycję na siponimod (Cmax i AUC), dlatego siponimod można przyjmować niezależnie od posiłków (patrz punkt 4.2).
Dystrybucja
Siponimod podlega dystrybucji do tkanek z umiarkowaną średnią objętością dystrybucji wynoszącą 124 litry. Frakcja siponimodu wykrywana w osoczu wynosi 68% u ludzi. Siponimod łatwo przenika przez barierę krew-mózg. Stopień wiązania siponimodu z białkami wynosi >99,9% u osób zdrowych oraz u pacjentów z zaburzeniami czynności wątroby lub nerek.
Metabolizm
Siponimod jest intensywnie metabolizowany, głównie przez cytochrom P450 2C9 (CYP2C9) (79,3%), a w mniejszym stopniu przez cytochrom P450 3A4 (CYP3A4) (18,5%).
Nie przewiduje się, aby działanie farmakologiczne głównych metabolitów M3 i M17 przyczyniało się do działania klinicznego oraz bezpieczeństwa stosowania siponimodu u ludzi.
Badania in vitro wykazały, że siponimod i jego główne metabolity ogólnoustrojowe M3 i M17 nie wykazują klinicznie istotnych interakcji lekowych po zastosowaniu dawki terapeutycznej wynoszącej 2 mg raz na dobę w odniesieniu do wszystkich badanych enzymów CYP i transporterów oraz nie wymagają badania klinicznego.
CYP2C9 jest enzymem polimorficznym, a jego genotyp wpływa na ułamkowy udział dwóch metabolicznych szlaków utleniania w całkowitej eliminacji leku. Modelowanie PBPK wskazuje na zróżnicowane hamowanie i indukcję szlaków CYP3A4 zależne od genotypu CYP2C9. Przy zmniejszonej aktywności metabolicznej CYP2C9 w odpowiednich genotypach przewiduje się większy wpływ enzymów modulujących CYP3A4 na ekspozycję na siponimod (patrz punkt 4.5).
Eliminacja
U pacjentów z SM oszacowano, że pozorny klirens układowy (CL/F) wynosi 3,11 l/h. Pozorny okres półtrwania siponimodu wynosi około 30 godzin.
Siponimod jest wydalany z krążenia układowego głównie w wyniku przemian metabolicznych i następującego po nich wydalania z żółcią/kałem. Siponimod w postaci niezmienionej nie był wykrywany w moczu.
Liniowość lub nieliniowość
Stężenie siponimodu zwiększa się w sposób proporcjonalny do dawki po wielokrotnym podawaniu dawek od 0,3 mg do 20 mg siponimodu raz na dobę.
Stężenia w osoczu w stanie stacjonarnym są osiągane po około 6 dniach podawania dawek leku raz na dobę, a stężenia w stanie stacjonarnym są około 2 do 3 razy większe niż po podaniu dawki początkowej. Aby osiągnąć dawkę terapeutyczną siponimodu wynoszącą 2 mg po 6 dniach, stosuje się schemat stopniowego zwiększania dawki, a do osiągnięcia stanu stacjonarnego w osoczu wymagane są dodatkowe 4 dni podawania leku.
Charakterystyka w szczególnych grupach pacjentów lub w szczególnych populacjach
Genotyp CYP2C9 Genotyp CYP2C9 wpływa na CL/F siponimodu. Dwie analizy farmakokinetyki populacyjnej wskazały, że pacjenci z genotypem CYP2C9*1*1 i *1*2 intensywnie metabolizują lek, pacjenci z genotypem *2*2 i *1*3 umiarkowanie metabolizują lek, a pacjenci z genotypem *2*3 i *3*3 słabo metabolizują lek. W porównaniu z osobami z genotypem CYP2C9*1*1, u osób z genotypami CYP2C9*2*2, *1*3, *2*3 i *3*3 wartości CL/F są mniejsze odpowiednio o 20%, 35-38%, 45-48% i 74%. Z tego względu ekspozycja na siponimod jest o około 25%, 61%, 91% i 284% większa u pacjentów z genotypem odpowiednio CYP2C9*2*2, *1*3, *2*3 i *3*3 w porównaniu z osobami z genotypem *1*1 (patrz tabela 4) (patrz punkty 4.2 i 4.4).
Istnieją inne, rzadziej występujące polimorfizmy CYP2C9. U takich pacjentów nie oceniano farmakokinetyki siponimodu. Niektóre polimorfizmy, takie jak *5, *6, *8 i *11, są związane ze zmniejszeniem lub utratą funkcji enzymów. Szacuje się, że allele CYP2C9*5, *6, *8 i *11 mają łączną częstość występowania wynoszacą około 10% w populacjach pochodzenia afrykańskiego, 2% u Latynosów / Hiszpanów i <0,4% u rasy białej i azjatyckiej.
Tabela 4 Wpływ genotypu CYP2C9 na CL/F i ogólnoustrojową ekspozycję na siponimod
|
Genotyp CYP2C9 |
Częstość występowania u osób rasy białej |
Szacunkowa wartość CL/F (l/h) |
% CL/F u osób z CYP2C9*1*1 |
% zwiększenie ekspozycji w porównaniu do CYP2C9*1*1 |
|
Pacjenci intensywnie metabolizujący lek |
||||
|
CYP2C9*1*1 |
62-65 |
3,1-3,3 |
100 |
- |
|
CYP2C9*1*1 |
20-24 |
3,1-3,3 |
99-100 |
- |
|
Pacjenci umiarkowanie metabolizujący lek |
||||
|
CYP2C9*2*2 |
1-2 |
2,5-2,6 |
80 |
25 |
|
CYP2C9*1*3 |
9-12 |
1,9-2,1 |
62-65 |
61 |
|
Pacjenci słabo metabolizujący lek |
||||
|
CYP2C9*2*3 |
1,4-1,7 |
1,6-1,8 |
52-55 |
91 |
|
CYP2C9*3*3 |
0,3-0,4 |
0,9 |
26 |
284 |
Pacjenci w podeszłym wieku
Wyniki farmakokinetyki populacyjnej sugerują, że nie ma konieczności dostosowania dawki u pacjentów w podeszłym wieku (w wieku 65 lat i starszych). Do badań klinicznych nie włączono pacjentów w wieku powyżej 61 lat. Siponimod należy stosować z zachowaniem ostrożności u pacjentów w podeszłym wieku (patrz punkt 4.2).
Płeć
Wyniki farmakokinetyki populacyjnej sugerują, że nie ma konieczności dostosowania dawki leku w zależności od płci.
Rasa, pochodzenie etniczne
Parametry farmakokinetyczne po podaniu pojedynczej dawki nie różniły się u osób zdrowych pochodzenia japońskiego i osób zdrowych rasy białej, co wskazuje na brak wpływu pochodzenia etnicznego pacjenta na farmakokinetykę siponimodu.
Zaburzenia czynności nerek
U pacjentów z łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek nie ma konieczności dostosowania dawki siponimodu. Średni okres półtrwania i Cmax siponimodu (stężenie całkowite i stężenie substancji niezwiązanej) były porównywalne u osób z ciężkimi zaburzeniami czynności nerek i u osób zdrowych. AUC całkowite i AUC substancji niezwiązanej były tylko nieznacznie zwiększone (o 23 do 33%) w porównaniu z osobami zdrowymi. Nie badano wpływu schyłkowej niewydolności nerek lub hemodializy na farmakokinetykę siponimodu. Ze względu na duży stopień wiązania siponimodu z białkami osocza (>99,9%), przewiduje się, że hemodializa nie zmieni całkowitego stężenia siponimodu i stężenia niezwiązanego siponimodu i w związku z tym, nie przewiduje się konieczności dostosowania dawki.
Zaburzenia czynności wątroby
Siponimodu nie wolno stosować u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkt 4.3). U pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności wątroby nie ma konieczności dostosowania dawki siponimodu. AUC niezwiązanego siponimodu jest o 15% i 50% większe odpowiednio u pacjentów z umiarkowanymi i ciężkimi zaburzeniami czynności wątroby w porównaniu z osobami zdrowymi w badaniu z zastosowaniem pojedynczej dawki 0,25 mg. Średni okres półtrwania siponimodu pozostał niezmieniony u osób z zaburzeniami czynności wątroby.
W badaniach toksyczności po podaniu wielokrotnym dawek leku u myszy, szczurów i małp siponimod znacząco wpływał na układ limfatyczny (limfopenia, atrofia tkanki limfoidalnej i zmniejszona odpowiedź przeciwciał), co jest zgodne z głównym działaniem farmakologicznym leku w receptorach S1P1 (patrz punkt 5.1).
Do działań toksycznych ograniczających dawkę u zwierząt należały działania nefrotoksyczne u myszy, rozwój masy ciała u szczurów oraz działania niepożądane na OUN i układ pokarmowy u małp. Głównymi narządami docelowymi dla działań toksycznych u gryzoni były płuca, wątroba, tarczyca, nerka oraz macica/pochwa. U małp dodatkowo obserwowano wpływ na mięśnie i skórę. Te działania toksyczne wystąpiły przy ogólnoustrojowym narażeniu na siponimod ponad 30-krotnie większym niż narażenie występujące u ludzi obliczane na podstawie AUC podczas stosowania dawki podtrzymującej wynoszącej 2 mg/dobę.
Siponimod nie miał działania fototoksycznego ani nie powodował uzależnienia i nie był genotoksyczny w warunkach in vitro i in vivo.
Działanie rakotwórcze
W badaniach rakotwórczości siponimod wywoływał chłoniaki, naczyniaki i naczyniakomięsaki krwionośne u myszy, natomiast u samców szczura stwierdzono występowanie gruczolaków pęcherzykowych i raków tarczycy. Występowanie tych guzów uznano za swoiste dla myszy lub przypisywano metabolicznym zmianom adaptacyjnym wątroby u szczególnie wrażliwych gatunków szczura; ich znaczenie dla ludzi jest niejasne.
Płodność i toksyczny wpływ na reprodukcję
Siponimod nie wpływał na płodność samców i samic szczura aż do największej badanej dawki, stanowiącej w przybliżeniu 19-krotność marginesu bezpieczeństwa w oparciu o ekspozycję ogólnoustrojową u ludzi (AUC) po podaniu dawki dobowej 2 mg.
Wiadomo, że receptor, na który działa siponimod (receptor sfingozyno-1-fosforanu) uczestniczy w powstawaniu naczyń krwionośnych podczas embriogenezy.
W badaniach dotyczących rozwoju zarodka i płodu przeprowadzonych u szczurów i królików, siponimod wywoływał działania embriotoksyczne przy braku toksycznego wpływu na matkę. U obu gatunków wzrosła śmiertelność przedurodzeniowa. Podczas gdy u szczurów odnotowano większą liczbę płodów z zewnętrznymi wadami wrodzonymi, wadami wrodzonymi kośćca i narządów trzewnych (np. rozszczep podniebienia i zniekształcone obojczyki, kardiomegalia i obrzęk), u płodów królika obserwowano głównie zmiany dotyczące kośćca i narządów trzewnych.
W badaniu rozwoju przed- i pourodzeniowego przeprowadzonym na szczurach stwierdzono zwiększoną liczbę martwego potomstwa (urodzonych martwych lub martwych przed 4. dniem po urodzeniu) lub młodych z wadami wrodzonymi (potomstwo płci męskiej z wadami wrodzonymi układu moczowo-płciowego i (lub) zmniejszeniem odległości anogenitalnej; młode obu płci z obrzękiem, opuchniętymi miękkimi kośćmi czaszki lub zgięciem kończyn tylnych).
Narażenie (AUC) przy odpowiednich dawkach NOAEL w odniesieniu do rozwoju zarodka i płodu (szczury i króliki) oraz rozwoju przed- i pourodzeniowego (szczury) było mniejsze od ogólnoustrojowego narażenia u ludzi (AUC) po podaniu dawki dobowej 2 mg i w związku z tym nie ma marginesu bezpieczeństwa.
Mayzent 0,25 mg tabletki powlekane
Rdzeń tabletki
Laktoza jednowodna
Celuloza mikrokrystaliczna
Krospowidon
Glicerolu dibehenian
Krzemionka koloidalna bezwodna
Otoczka tabletki
Alkohol poliwinylowy
Tytanu dwutlenek (E171)
Żelaza tlenek czerwony (E172)
Żelaza tlenek czarny (E172)
Talk
Lecytyna sojowa
Guma ksantanowa
Mayzent 1 mg tabletki powlekane
Rdzeń tabletki
Laktoza jednowodna
Celuloza mikrokrystaliczna
Krospowidon
Glicerolu dibehenian
Krzemionka koloidalna bezwodna
Otoczka tabletki
Alkohol poliwinylowy
Tytanu dwutlenek (E171)
Żelaza tlenek czerwony (E172)
Żelaza tlenek czarny (E172)
Talk
Lecytyna sojowa
Guma ksantanowa
Mayzent 2 mg tabletki powlekane
Rdzeń tabletki
Laktoza jednowodna
Celuloza mikrokrystaliczna
Krospowidon
Glicerolu dibehenian
Krzemionka koloidalna bezwodna
Otoczka tabletki
Alkohol poliwinylowy
Tytanu dwutlenek (E171)
Żelaza tlenek żółty (E172)
Żelaza tlenek czerwony (E172)
Talk
Lecytyna sojowa
Guma ksantanowa
Nie dotyczy.
2 lata
Nie przechowywać w temperaturze powyżej 25°C.
Mayzent 0,25 mg tabletki powlekane
Opakowania przeznaczone do zwiększania dawki zawierające 12 tabletek powlekanych w blistrach
z PA/Aluminium/PVC/Aluminium w etui.
Opakowania zawierające 84 lub 120 tabletek powlekanych w blistrach
z PA/Aluminium/PVC/Aluminium.
Mayzent 1 mg tabletki powlekane
Opakowania zawierające 28 lub 98 tabletek powlekanych w blistrach
z PA/Aluminium/PVC/Aluminium.
Mayzent 2 mg tabletki powlekane
Opakowania zawierające 14, 28 lub 98 tabletek powlekanych w blistrach
z PA/Aluminium/PVC/Aluminium.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Novartis Europharm Limited
Vista Building
Elm Park,
Merrion Road
Dublin 4
Irlandia
Mayzent 0,25 mg tabletki powlekane
EU/1/19/1414/001
EU/1/19/1414/002
EU/1/19/1414/004
Mayzent 1 mg tabletki powlekane
EU/1/19/1414/007
EU/1/19/1414/008
Mayzent 2 mg tabletki powlekane
EU/1/19/1414/003
EU/1/19/1414/005
EU/1/19/1414/006
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 13 stycznia 2020
Data ostatniego przedłużenia pozwolenia: 19 września 2024
Szczegółowe informacje o tym produkcie leczniczym są dostępne na stronie internetowej Europejskiej Agencji Leków https://www.ema.europa.eu.
Powered by Biogen Poland Sp. z o.o.
(Biogen-101710)
 i
i  "Dodaj aplikację do ekranu początkowego"
"Dodaj aplikację do ekranu początkowego"