Powered by Biogen Poland Sp. z o.o.
Powered by Biogen Poland Sp. z o.o.
Ocrevus 300 mg koncentrat do sporządzania roztworu do infuzji
Każda fiolka zawiera 300 mg okrelizumabu w 10 ml w stężeniu 30 mg/ml. Końcowe stężenie produktu leczniczego po rozcieńczeniu wynosi około 1,2 mg/ml.
Okrelizumab jest humanizowanym przeciwciałem monoklonalnym, wytwarzanym w komórkach jajnika chomika chińskiego w technologii rekombinacji DNA.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Koncentrat do sporządzania roztworu do infuzji.
Roztwór przejrzysty do lekko opalizującego, bezbarwny do jasnobrązowego.
Produkt leczniczy Ocrevus jest wskazany w leczeniu dorosłych pacjentów z rzutowymi postaciami stwardnienia rozsianego (ang. relapsing forms of multiple sclerosis, RMS), z aktywną chorobą, definiowaną na podstawie cech klinicznych lub radiologicznych (patrz punkt 5.1).
Produkt leczniczy Ocrevus jest wskazany w leczeniu dorosłych pacjentów z wczesną pierwotnie postępującą postacią stwardnienia rozsianego (ang. primary progressive multiple sclerosis, PPMS) ocenianą na podstawie czasu trwania choroby i poziomu niesprawności, a także cech radiologicznych charakterystycznych dla aktywności zapalnej (patrz punkt 5.1).
Leczenie powinno być rozpoczynane i nadzorowane przez lekarzy specjalistów mających doświadczenie w diagnozowaniu i leczeniu stanów neurologicznych, posiadających dostęp do odpowiednich środków medycznych niezbędnych w leczeniu ciężkich reakcji, takich jak ciężkie reakcje związane z wlewem (ang. infusion related reactions, IRRs).
Premedykacja zapobiegająca reakcjom związanym z wlewem
Przed każdym podaniem okrelizumabu konieczne jest zastosowanie następujących dwóch rodzajów premedykacji, aby zmniejszyć częstotliwość i nasilenie reakcji związanych z wlewem (dalsze kroki mające na celu zmniejszyć występowanie reakcji związanych z wlewem, patrz punkt 4.4):
Dodatkowo można również rozważyć premedykację lekiem przeciwgorączkowym (np. paracetamolem) na około 30-60 minut przed każdym podaniem wlewu.
Dawkowanie:
Dawka początkowa
Początkową dawkę 600 mg podaje się w dwóch oddzielnych wlewach dożylnych; najpierw wlew 300 mg, a 2 tygodnie później drugi wlew 300 mg (patrz Tabela 1).
Kolejne dawki
Kolejne dawki okrelizumabu to pojedynczy wlew dożylny dawki 600 mg podawany co 6 miesięcy (patrz Tabela 1). Pierwszą kolejną dawkę 600 mg należy podać sześć miesięcy po pierwszym wlewie dawki początkowej.
Pomiędzy kolejnymi dawkami okrelizumabu należy zachować odstęp minimum 5 miesięcy.
Dostosowanie podania w przypadku wystąpienia reakcji związanych z wlewem
Zagrażające życiu reakcje związane z wlewem
Jeżeli w czasie podania wlewu wystąpią objawy świadczące o reakcji związanej z wlewem stanowiącej zagrożenie życia lub powodującej niesprawność, takie jak ostra nadwrażliwość lub zespół ostrej niewydolności oddechowej, podanie należy natychmiast przerwać, a pacjentowi należy zapewnić odpowiednie leczenie. Wlew u tych pacjentów należy zakończyć i nie należy go wznawiać (patrz punkt 4.3).
Ciężkie reakcje związane z wlewem
Jeżeli u pacjenta wystąpi ciężka reakcja związana z wlewem (taka jak duszność) lub zespół następujących objawów: zaczerwienienie twarzy, gorączka i ból gardła, należy natychmiast przerwać podanie wlewu oraz zastosować leczenie objawowe. Podanie wlewu należy wznowić dopiero po ustąpieniu wszystkich objawów. Podanie wlewu należy wznowić z szybkością równą połowie szybkości w chwili wystąpienia reakcji. Dostosowywanie podania wlewu nie jest konieczne w przypadku kolejnych nowych podań wlewu, o ile u pacjenta nie wystąpi reakcja związana z wlewem.
Reakcje związane z wlewem o nasileniu łagodnym do umiarkowanego
Jeżeli u pacjenta wystąpi łagodna lub umiarkowana reakcja związana z wlewem (np. bóle głowy), szybkość podania wlewu należy zmniejszyć do połowy szybkości podania wlewu w chwili
wystąpienia zdarzenia. Zmniejszona szybkość powinna zostać utrzymana przez przynajmniej 30 minut. W przypadku tolerancji, szybkość podania wlewu można następnie zwiększyć zgodnie z początkową prędkością podania wlewu stosowaną u pacjenta. Dostosowywanie podania wlewu nie jest wymagane w przypadku kolejnych nowych podań wlewu, o ile u pacjenta nie wystąpi reakcja związana z
wlewem.
Modyfikacja dawki w trakcie leczenia
Powyższe przykłady przerwania podawania produktu lub zmniejszenia szybkości wlewu (z powodu łagodnych, umiarkowanych lub ciężkich reakcji związanych z wlewem) spowodują zmianę prędkości podania wlewu oraz wydłużą całkowity czas trwania podania wlewu, ale nie zwiększą całkowitej dawki.
Zmniejszanie dawki nie jest zalecane.
Opóźnienie przyjęcia dawki lub pominięcie dawki
W przypadku pominięcia podania wlewu, należy go podać tak szybko, jak to możliwe; nie należy czekać do terminu kolejnej zaplanowanej dawki. Pomiędzy dawkami należy zachować odstęp 6 miesięcy (minimum 5 miesięcy) (patrz Tabela 1).
Szczególne populacje pacjentów
Dorośli w wieku powyżej 55 lat
Na podstawie dostępnych ograniczonych danych (patrz punkty 5.1 i 5.2) nie ma potrzeby dostosowywania dawkowania u pacjentów w wieku powyżej 55 lat. Pacjenci włączeni do trwających badań klinicznych kontynuują przyjmowanie dawki 600 mg okrelizumabu co sześć miesięcy po przekroczeniu wieku 55 lat.
Zaburzenia czynności nerek
Nie przeprowadzono formalnych badań bezpieczeństwa stosowania i skuteczności okrelizumabu u pacjentów z zaburzeniami czynności nerek. Pacjenci z łagodnymi zaburzeniami czynności nerek byli włączeni do badań klinicznych. Brak doświadczenia u pacjentów z umiarkowanymi i ciężkimi zaburzeniami czynności nerek. Okrelizumab jest przeciwciałem monoklonalnym usuwanym na drodze katabolizmu (tj. rozpadu na peptydy i aminokwasy), dlatego dostosowanie dawkowania nie wydaje się konieczne u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2).
Zaburzenia czynności wątroby
Nie przeprowadzono formalnych badań bezpieczeństwa stosowania i skuteczności okrelizumabu u pacjentów z zaburzeniami czynności wątroby. Pacjenci z łagodnymi zaburzeniami czynności wątroby byli włączeni do badań klinicznych. Brak doświadczenia u pacjentów z umiarkowanymi i ciężkimi zaburzeniami czynności wątroby. Okrelizumab jest przeciwciałem monoklonalnym usuwanym na drodze katabolizmu (a nie metabolizmu wątrobowego), dlatego dostosowanie dawkowania nie wydaje się konieczne w przypadku pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.2).
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania i skuteczności okrelizumabu u dzieci i młodzieży w wieku od 0 do 18 lat. Dane nie są dostępne.
Sposób podawania
Produkt leczniczy Ocrevus 300 mg koncentrat do sporządzania roztworu do infuzji nie jest przeznaczony do podskórnego podawania i powinien być podawany wyłącznie we wlewie dożylnym.
Ważne jest, by sprawdzić etykiety produktu i upewnić się, że pacjentowi jest podawany produkt w odpowiedniej postaci (dożylnej lub podskórnej), zgodnie z zaleceniem lekarza.
Pacjenci mogą rozpocząć leczenie okrelizumabem w postaci dożylnej lub podskórnej.
Po rozcieńczeniu, leczenie jest podawane we wlewie dożylnym przez oddzielną linię infuzyjną. Wlewu nie należy podawać w postaci wstrzyknięcia dożylnego lub bolusa.
Jeśli u pacjenta nie wystąpiła ciężka reakcja związana z wlewem (IRR) podczas żadnego z wcześniejszych wlewów okrelizumabu, kolejne dawki można podać w krótszym (2-godzinnym) wlewie ( patrz Tabela 1, Opcja 2).
Tabela 1: Dawkowanie i schemat podawania
|
|
Ilość okrelizumabu, którą należy podać |
Wskazówki dotyczące podania wlewu |
|
|
Dawka początkowa (600 mg) podzielona na 2 wlewy |
Wlew 1 |
300 mg w 250 ml |
180 ml/godzinę
|
|
Wlew 2 (2 tygodnie później) |
300 mg w 250 ml |
||
|
|
|
600 mg w 500 ml |
200 ml/godzinę
|
|
|
Opcja 1 |
|
|
|
|
Wlew trwający |
|
|
|
|
około 3,5 godziny |
|
|
|
Kolejne dawki (600mg) pojedynczy wlew raz na 6 miesięcy |
|
|
|
|
LUB |
|||
|
|
|
600 mg w 500 ml |
|
|
|
Opcja 2 |
|
|
|
|
Wlew trwający |
|
|
|
|
około 2 godzin |
|
|
Roztwory do wlewów dożylnych przygotowuje się, rozcieńczając koncentrat w worku infuzyjnym zawierającym roztwór chlorku sodu do infuzji o stężeniu 9 mg/ml (0,9%), do uzyskania końcowego stężenia okrelizumabu wynoszącego około 1,2 mg/ml.
Instrukcje dotyczące rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6.
Pacjentów należy monitorować w trakcie podawania wlewu i przez co najmniej jedną godzinę po zakończeniu wlewu (patrz punkt 4.4).
Identyfikowalność
W celu poprawy identyfikowalności biologicznych produktów leczniczych, nazwa handlowa oraz numer serii podanego produktu powinny być wyraźnie zapisane w dokumentacji pacjenta.
Reakcje związane z wlewem (IRR)
Podawanie okrelizumabu wiąże się z występowaniem reakcji związanych z wlewem, które mogą być związane z uwalnianiem cytokin i (lub) innych mediatorów reakcji chemicznych.
Objawy reakcji związanych z wlewem mogą wystąpić w trakcie każdego podania wlewu okrelizumabu, ale częściej zgłaszano je w trakcie pierwszego podania wlewu. Reakcje związane z wlewem mogą wystąpić w ciągu 24 godzin od podania (patrz punkt 4.8). Reakcje tego typu mogą mieć postać świądu, wysypki, pokrzywki, rumienia, podrażnienia gardła, bólu jamy ustnej i gardła, duszności, obrzęku gardła lub krtani, zaczerwienienia twarzy, hipotensji, gorączki, zmęczenia, bólu głowy, zawrotów głowy, nudności, częstoskurczu i anafilaksji.
Przed podaniem wlewu
Postępowanie z ciężkimi reakcjami
Należy zapewnić dostęp do odpowiednich środków i sprzętu niezbędnego w postępowaniu z ciężkimi reakcjami, takimi jak ciężkie reakcje związane z wlewem, reakcje nadwrażliwości i (lub) reakcje
anafilaktyczne.
Niedociśnienie
W trakcie podawania wlewu okrelizumabu może wystąpić niedociśnienie tętnicze jako objaw reakcji związanej z wlewem. Dlatego należy rozważyć odstawienie leków przeciwnadciśnieniowych na 12
godzin przed i podczas każdego podania wlewu. Pacjenci z zastoinową niewydolnością serca (klasa III i IV według New York Heart Association) w wywiadzie, nie brali udziału w badaniach.
Premedykacja
Pacjenci muszą otrzymać premedykację, aby zmniejszyć częstość i nasilenie reakcji związanych z podaniem wlewu (patrz punkt 4.2).
W trakcie podawania wlewu
Należy podjąć następujące kroki u pacjentów, u których wystąpią ciężkie objawy ze strony płuc, takie jak skurcz oskrzeli lub zaostrzenie astmy:
Objawy reakcji nadwrażliwości mogą być klinicznie niemożliwe do odróżnienia od objawów reakcji związanej z wlewem. Jeżeli zachodzi podejrzenie wystąpienia reakcji nadwrażliwości podczas podawania wlewu, należy natychmiast i na stałe przerwać podanie wlewu (patrz „Reakcje nadwrażliwości” poniżej).
Po podaniu wlewu
Pacjentów należy obserwować przez przynajmniej jedną godzinę po zakończeniu podania wlewu w celu wykrycia objawów reakcji związanej z wlewem.
Lekarze powinni ostrzec pacjentów, że reakcja związana z wlewem może wystąpić w ciągu 24 godzin od zakończenia podawania wlewu.
Wskazówki dotyczące dostosowania wlewu w przypadku wystąpienia reakcji związanych z wlewem, patrz punkt 4.2.
Reakcje nadwrażliwości
Może również wystąpić reakcja nadwrażliwości (ostra reakcja alergiczna na produkt leczniczy). Ostre reakcje nadwrażliwości typu 1 (IgE-zależne) mogą być klinicznie niemożliwe do odróżnienia od objawów reakcji związanych z wlewem.
Reakcja nadwrażliwości może rozwinąć się w trakcie każdego podania produktu, chociaż zazwyczaj nie występuje w trakcie pierwszego podania. Jeżeli w trakcie kolejnych podań wystąpią objawy cięższe niż poprzednio lub jeśli wystąpią nowe ciężkie objawy, należy założyć podejrzenie wystąpienia reakcji nadwrażliwości. Nie należy stosować leczenia u pacjentów ze znaną nadwrażliwością IgE-zależną na okrelizumab lub na którąkolwiek z substancji pomocniczych (patrz punkt 4.3).
Zakażenie
Podawanie okrelizumabu musi być opóźnione u pacjentów z aktywnym zakażeniem do czasu ustąpienia tego zakażenia.
Zaleca się ocenę stanu układu immunologicznego pacjenta przed podaniem produktu leczniczego, ponieważ pacjenci z ciężkim obniżeniem odporności (np. z limfopenią, neutropenią, hipogammaglobulinemią) nie powinni być leczeni (patrz punkty 4.3 i 4.8).
Całkowity odsetek pacjentów, u których wystąpiło ciężkie zakażenie był podobny, jak po podaniu produktów porównywanych w badaniu (patrz punkt 4.8). Częstość występowania zakażeń stopnia 4. (zagrażających życiu) i stopnia 5. (śmiertelnych) była niska we wszystkich grupach poddanych leczeniu, jednak w przypadku PPMS częstość występowania zakażeń zagrażających życiu (1,6% w porównaniu z 0,4%) i śmiertelnych (0,6% w porównaniu z 0%) była większa po zastosowaniu okrelizumabu niż placebo. Wszystkie zakażenia zagrażające życiu ustąpiły bez konieczności zaprzestania podawania okrelizumabu.
W PPMS, pacjenci z trudnościami w przełykaniu podlegają wyższemu ryzyku aspiracyjnego zapalenia płuc. Leczenie okrelizumabem może dodatkowo zwiększać ryzyko ciężkiego zapalenia płuc u tych pacjentów. Lekarze powinni podjąć niezwłoczne działania w przypadku pacjentów z objawami zapalenia płuc.
Postępująca wieloogniskowa leukoencefalopatia (PML)
Zakażenie wirusem Johna Cunninghama (JCV) powodującym PML obserwowano bardzo rzadko u pacjentów leczonych przeciwciałami anty-CD20, w tym okrelizumabem i było ono przeważnie związane z czynnikami ryzyka (populacja pacjentów np. z limfopenią, w podeszłym wieku, politerapia lekami immunosupresyjnymi).
Lekarze powinni zachować czujność wobec wczesnych przedmiotowych i podmiotowych objawów PML, do których należą wszelkie nowe objawy lub nasilenie już istniejących przedmiotowych i podmiotowych objawów neurologicznych, ponieważ mogą one przypominać stwardnienie rozsiane.
W przypadku podejrzenia PML należy wstrzymać podawanie okrelizumabu. Należy rozważyć przeprowadzenie oceny, w tym wykonanie badania rezonansem magnetycznym (MRI), najlepiej z kontrastem (wynik należy porównać z wynikiem sprzed leczenia), badanie płynu mózgowo-rdzeniowego w kierunku obecności kwasu deoksyrybonukleinowego (DNA) JCV oraz powtarzane badania neurologiczne. Jeżeli rozpoznanie PML zostanie potwierdzone, leczenie należy przerwać i nie wznawiać.
Reaktywacja wirusowego zapalenia wątroby typu B
Reaktywacja wirusa zapalenia wątroby typu B (ang. hepatitis B virus, HBV), w niektórych przypadkach prowadząca do piorunującego zapalenia wątroby, niewydolności wątroby i zgonu, była zgłaszana u pacjentów leczonych przeciwciałami anty-CD20.
Przed rozpoczęciem leczenia u wszystkich pacjentów należy wykonać badania przesiewowe w kierunku HBV, zgodnie z lokalnymi wytycznymi. Pacjentów z czynnym wirusem HBV (tj. aktywne zakażenie potwierdzone dodatnimi wynikami badań na obecność antygenu powierzchniowego wirusa HBV (HBsAg) i przeciwciał anty-HB) nie należy leczyć okrelizumabem (patrz punkt 4.3). Pacjentów z dodatnim wynikiem badań serologicznych (tj. ujemny wynik HBsAg i dodatni wynik przeciwciała przeciwko antygenowi rdzeniowemu wirusa HBV (HBcAg+), nosiciele HBV (obecność antygenu powierzchniowego, HBsAg+) należy skonsultować przez specjalistów chorób wątroby przed rozpoczęciem leczenia. Pacjentów tych należy monitorować i leczyć zgodnie z lokalnymi standardami medycznymi, aby zapobiec reaktywacji wirusowego zapalenia wątroby typu B.
Późna neutropenia
Zgłaszano przypadki neutropenii o późnym początku występującej po co najmniej 4 tygodniach od podania ostatniego wlewu okrelizumabu (patrz punkt 4.8). Chociaż niektóre przypadki miały nasilenie stopnia 3. lub 4., większość przypadków była stopnia 1. lub 2. U pacjentów z przedmiotowymi i podmiotowymi objawami zakażenia zaleca się oznaczanie liczby granulocytów obojętnochłonnych we krwi.
Nowotwory złośliwe
W kontrolowanym okresie rejestracyjnych badań klinicznych obserwowano zwiększoną liczbę nowotworów złośliwych (w tym raka piersi) u pacjentów leczonych okrelizumabem, w porównaniu z grupami kontrolnymi. Zapadalność mieściła się w przedziale wartości przewidywanych dla populacji pacjentów ze stwardnieniem rozsianym. Po około 10 latach ciągłego leczenia okrelizumabem w okresie kontrolowanym i w otwartej fazie przedłużonej (ang. Open-Label Extension, OLE) rejestracyjnych badań klinicznych zapadalność na nowotwory złośliwe pozostała w przedziale wartości przewidywanych dla populacji pacjentów z SM. Pacjentów ze znanym aktywnym nowotworem złośliwym nie należy leczyć
okrelizumabem (patrz punkt 4.3). Należy dokonać indywidualnej oceny stosunku korzyści do ryzyka u pacjentów ze znanymi czynnikami ryzyka wystąpienia nowotworów złośliwych oraz u pacjentów, którzy są aktywnie monitorowani ze względu na ryzyko nawrotu nowotworu złośliwego. Pacjenci powinni poddać się standardowym badaniom przesiewowym w kierunku raka piersi, zgodnie z lokalnymi wytycznymi.
Leczenie pacjentów z ciężkim osłabieniem odporności
Pacjenci w stanie ciężkiego obniżenia odporności nie mogą otrzymywać leczenia do czasu ustąpienia tego stanu (patrz punkt 4.3).
W innych chorobach autoimmunologicznych stosowanie okrelizumabu jednocześnie z lekami immunosupresyjnymi (np. przewlekłe podawanie kortykosteroidów, niebiologicznych i biologicznych leków przeciwreumatycznych modyfikujących przebieg choroby (ang. biologic disease- modifying antirheumatic drugs, DMARDS), mykofenolanu mofetylu, cyklofosfamidu, azatiopryny) powodowało zwiększenie przypadków ciężkich zakażeń, w tym zakażeń oportunistycznych. Zakażenia obejmowały m.in. atypowe zapalenie płuc, zapalenie płuc wywołane przez Pneumocystis jirovecii, zapalenia płuc w przebiegu ospy wietrznej, gruźlicę, histoplazmozę. W rzadkich przypadkach niektóre z tych zakażeń były śmiertelne. W analizie eksploracyjnej zidentyfikowano następujące czynniki związane z ryzykiem ciężkich zakażeń: wyższe niż rekomendowane w stwardnieniu rozsianym dawki okrelizumabu, inne choroby współistniejące i przewlekłe stosowanie leków immunosupresyjnych/kortykosteroidów.
Nie zaleca się stosowania innych leków immunosupresyjnych w skojarzeniu z okrelizumabem, poza kortykosteroidami w objawowym leczeniu rzutów. Wiedza o tym, czy jednoczesne stosowanie steroidów w objawowym leczeniu rzutów wiąże się ze zwiększonym ryzykiem zakażeń w praktyce klinicznej, jest ograniczona. W badaniach rejestracyjnych z zastosowaniem okrelizumabu w stwardnieniu rozsianym podawanie kortykosteroidów w leczeniu rzutu nie wiązało się ze zwiększonym ryzykiem ciężkiego zakażenia.
Rozpoczynając podawanie okrelizumabu po leczeniu immunosupresyjnym lub rozpoczynając leczenie immunosupresyjne po leczeniu okrelizumabem należy wziąć pod uwagę prawdopodobieństwo nakładania się efektów farmakodynamicznych (patrz punkt 5.1, Właściwości farmakodynamiczne). Należy zachować ostrożność, przepisując okrelizumab, biorąc pod uwagę farmakodynamikę innych leków modyfikujących przebieg stwardnienia rozsianego.
Szczepienia
Nie przeprowadzono badań bezpieczeństwa immunizacji szczepionkami żywymi lub żywymi atenuowanymi po zakończeniu leczenia okrelizumabem i podawanie szczepionek zawierających żywe lub żywe atenuowane wirusy nie jest rekomendowane w trakcie leczenia oraz do czasu odnowy limfocytów B. W badaniach klinicznych mediana czasu do odnowy limfocytów B wyniosła 72 tygodnie (patrz punkt 5.1).
W randomizowanym otwartym badaniu pacjenci z RMS byli w stanie uzyskać odpowiedzi humoralne, choć były one słabsze, na toksoid tężcowy, 23-walentną polisacharydową szczepionkę przeciwko pneumokokom (23-PPV) ze szczepionką przypominającą lub bez niej, neoantygen hemocyjaniny (ang. keyhole limpet hemocyanin, KLH) i szczepionki przeciwko grypie sezonowej (patrz punkt 4.5 i 5.1).
Zaleca się szczepienie pacjentów leczonych okrelizumabem inaktywowanymi szczepionkami przeciwko grypie sezonowej.
Lekarze powinni sprawdzać status szczepień pacjentów, u których rozważają leczenie okrelizumabem. Pacjenci, którzy wymagają podania szczepionek powinni ukończyć szczepienia przynajmniej 6 tygodni przed rozpoczęciem leczenia.
Narażenie in utero na okrelizumab a szczepienie noworodków i niemowląt szczepionkami żywymi lub żywymi atenuowanymi
Ze względu na potencjalną deplecję limfocytów B u niemowląt matek, które były narażone na działanie okrelizumabu w okresie ciąży, zaleca się, by szczepienia szczepionkami żywymi lub żywymi atenuowanymi były opóźnione do czasu powrotu liczby limfocytów B do normy; z tego względu zaleca się pomiar liczby CD19-dodatnich limfocytów B u noworodków i niemowląt przed szczepieniem.
Zaleca się, by wszystkie szczepienia inne niż szczepienia szczepionkami żywymi lub żywymi atenuowanymi były podawane zgodnie z lokalnie obowiązującym kalendarzem szczepień oraz należy rozważyć oznaczanie miana odpowiedzi na szczepienie, aby sprawdzić, czy u danej osoby wzrosła ochronna odpowiedź immunologiczna, ponieważ skuteczność szczepienia może być zmniejszona.
Bezpieczeństwo i termin szczepienia należy omówić z lekarzem prowadzącym niemowlę (patrz punkt 4.6).
Sód
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, to znaczy lek uznaje się za „wolny od sodu”.
Nie przeprowadzono badań interakcji, ponieważ nie przewiduje się wystąpienia żadnych interakcji za pośrednictwem enzymów cytochromu P450, innych enzymów metabolizujących lub transporterów.
Szczepienia
Nie przeprowadzono badań nad bezpieczeństwem immunizacji szczepionkami żywymi lub żywymi atenuowanymi po zakończeniu leczenia okrelizumabem.
Dostępne są dane o wpływie toksoidu tężcowego, 23-walentnej polisacharydowej szczepionki przeciwko pneumokokom, neoantygenu KLH i szczepionki przeciwko grypie sezonowej na pacjentów przyjmujących okrelizumab (patrz punkt 4.4 i 5.1).
W okresie 2 lat od zakończenia leczenia odsetek pacjentów z dodatnim mianem przeciwciał przeciwko S. pneumoniae, śwince, różyczce i ospie wietrznej był podobny do wartości wyjściowych.
Leki immunosupresyjne
Nie zaleca się stosowania innych terapii immunosupresyjnych jednocześnie z okrelizumabem, z wyjątkiem kortykosteroidów podawanych w objawowym leczeniu rzutów (patrz punkt 4.4).
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym powinny stosować antykoncepcję w trakcie leczenia okrelizumabem oraz przez 4 miesiące od ostatniego podania dawki okrelizumabu.
Ciąża
Dane na temat stosowania okrelizumabu u kobiet w ciąży są ograniczone. Okrelizumab jest immunoglobuliną G (IgG). Wiadomo, że IgG przenika przez barieręłożyskową. Należy rozważyć opóźnienie szczepienia szczepionkami żywymi lub żywymi atenuowanymi noworodków i niemowląt, których matki w trakcie życia płodowego tych dzieci były narażone na okrelizumab. Nie gromadzono danych o liczbie limfocytów B u niemowląt narażonych na działanie okrelizumabu, a potencjalny czas trwania deplecji limfocytów B u noworodków i niemowląt jest nieznany (patrz punkt 4.4).
Zgłaszano przypadki przemijającego zmniejszenia liczby limfocytów B we krwi obwodowej oraz limfocytopenii u niemowląt kobiet poddanych ekspozycji na inne przeciwciała anty-CD20 w okresie ciąży. Deplecję limfocytów B in utero wykryto także w badaniach na zwierzętach.
Badania na zwierzętach (toksyczności dla zarodka i płodu) nie wykazały działania teratogennego. W badaniach nad rozwojem w okresie przed- i pourodzeniowym obserwowano toksyczny wpływ na reprodukcję (patrz punkt 5.3).
Należy unikać stosowania okrelizumabu w okresie ciąży, chyba że potencjalne korzyści dla matki przewyższają potencjalne ryzyko dla płodu.
Karmienie piersią
Wiadomo, że ludzkie IgG przenikają do mleka kobiecego w pierwszych kilku dniach po urodzeniu (okres wytwarzania siary), a wkrótce potem ich stężenie zmniejsza się do małych wartości.
W prospektywnym, wieloośrodkowym, otwartym badaniu MN42989 (SOPRANINO) 13 kobietom karmiącym piersią podano okrelizumab po medianie 2,0 miesiąca od porodu (zakres 0,5-5,0 miesiąca). Małe stężenia okrelizumabu były wykrywane w mleku kobiecym w okresie 60 dni po pierwszym wlewie podanym matce po porodzie (mediana względnej dawki u niemowlęcia wyniosła 0,27% [zakres 0,0-1,8%]), co wskazuje na minimalne przenikanie okrelizumabu do mleka kobiecego. Po 30 dniach od pierwszego wlewu podanego matce po porodzie okrelizumab był niewykrywalny we wszystkich dostępnych próbkach surowicy pobranych od niemowląt karmionych piersią (n=9), a stężenie limfocytów B u niemowląt mieściło się w granicach normy we wszystkich dostępnych próbkach krwi (n=10). Nie obserwowano wpływu okrelizumabu na stan zdrowia, wzrost i rozwój niemowląt karmionych piersią przez matki leczone w okresie obserwacji trwającym 44,6 tygodnia (zakres 8,6-62,7 tygodnia).
Chociaż nie ma dostępnych danych klinicznych dotyczących niemowląt potencjalnie narażonych na działanie okrelizumabu poprzez mleko matki, które otrzymują szczepionki żywe lub atenuowane, nie przewiduje się ryzyka ze względu na normalny poziom komórek B oraz niewykrywalny poziom okrelizumabu w surowicy obserwowany u tych niemowląt.
Małe stężenia okrelizumabu w mleku kobiecym (mediana względnej dawki u niemowląt wyniosła 0,1% [zakres 0,07-0,7%]) w okresie 90 dni po pierwszym wlewie podanym matce po porodzie były także obserwowane u 29 karmiących kobiet, którym podano okrelizumab po medianie 4,3 miesiąca (zakres 0,1-36 miesięcy) po porodzie w odrębnym prospektywnym badaniu klinicznym. W okresie obserwacji 21 niemowląt karmionych piersią przez co najmniej 2 tygodnie obserwowano ich prawidłowy wzrost i rozwój do 1 roku życia.
Okrelizumab może być stosowany w okresie karmienia piersią począwszy od kilku dni po porodzie.
Płodność
Dane przedkliniczne nie wykazują wyraźnego ryzyka dla ludzi na podstawie badań płodności samców i samic małp cynomolgus.
Produkt leczniczy Ocrevus nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
W kontrolowanym okresie rejestracyjnych badań klinicznych najważniejszymi i najczęściej zgłaszanymi działaniami niepożądanymi były reakcje związane z wlewem(34,3%, 40,1% odpowiednio w RMS i PPMS) oraz zakażenia (58,5%, 72,2% odpowiednio w
RMS i PPMS) (patrz punkt 4.4).
W kontrolowanym okresie rejestracyjnych badań klinicznych uczestniczyło łącznie 2376 pacjentów; 1852 spośród nich zostało włączonych do fazy OLE. Wszyscy pacjenci zmienili leczenie na okrelizumab w fazie OLE. Fazę OLE ukończyło 1155 pacjentów, co daje około 10 lat ciągłego leczenia okrelizumabem (15515 pacjento-lat ekspozycji) w kontrolowanym okresie badań i w fazie OLE. Ogólny profil bezpieczeństwa obserwowany w okresie leczenia kontrolowanego i w fazie OLE pozostaje spójny z ogólnym profilem bezpieczeństwa obserwowanym w okresie leczenia kontrolowanego.
Tabelaryczne zestawienie działań niepożądanych
W Tabeli 2 poniżej wymieniono działania niepożądane zgłaszane w kontrolowanym okresie rejestracyjnych badań klinicznych i pochodzące ze zgłoszeń spontanicznych. Te działania niepożądane zostały wymienione według klasyfikacji układów i narządów MedDRA oraz według częstości występowania. Częstość występowania określano w następujący sposób: bardzo często (≥ 1/10), często (≥ 1/100 do <1/10), niezbyt często (≥ 1/1 000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1 000), bardzo rzadko (<1/10 000) oraz częstość nieznana (częstość nie może być określona na podstawie dostępnych danych). W każdej grupie układów i narządów działania niepożądane wymieniono zgodnie ze zmniejszającą się częstością.
Tabela 2: Działania niepożądane
|
MedDRA
Klasyfikacja układów i narządów |
Bardzo często |
Często |
Częstość nieznana2 |
|
Zakażenia i zarażeniapasożytnicze |
Zakażenie górnych dróg oddechowych, zapalenie nosogardła, grypa |
Zapalenie zatok, zapalenie oskrzeli, opryszczka jamy ustnej, zapalenie żołądka i jelit, zakażenie układu oddechowego, zakażenie wirusowe, półpasiec, zapalenie spojówek, zapalenie tkanki łącznej |
|
|
Zaburzenia krwi i układu chłonnego
|
|
Neutropenia |
Neutropenia o późnym początku2 |
|
Zaburzenia układu oddechowego, klatkipiersiowej i śródpiersia |
|
Kaszel, nieżyt błony śluzowej nosa |
|
|
Badania diagnostyczne |
Zmniejszone stężenie immunoglobuliny M we krwi |
Zmniejszone stężenie immunoglobuliny G we krwi |
|
|
Urazy, zatrucia i powikłania po zabiegach |
Reakcja związana z wlewem1 |
|
|
1 Patrz opis wybranych działań niepożądanych.
2 Obserwowane po wprowadzeniu produktu do obrotu.
Opis wybranych działań niepożądanych
Reakcje związane z wlewem
W badaniach dotyczących RMS i PPMS objawy reakcji związanych z wlewem obejmowały między innymi: świąd, wysypkę, pokrzywkę, rumień, zaczerwienienie twarzy, niedociśnienie, gorączkę, zmęczenie, ból głowy, zawroty głowy, podrażnienie gardła, ból części ustnej gardła, duszność, obrzęk gardła lub krtani, nudności, częstoskurcz. W badaniach z grupą kontrolną nie obserwowano śmiertelnych reakcji związanych z wlewem. Dodatkowo objawy reakcji związanych z wlewem w okresie po wprowadzeniu produktu do obrotu obejmowały anafilaksję.
W badaniach klinicznych kontrolowanych substancją czynną (RMS) reakcja związana z wlewem była najczęściej występującym działaniem niepożądanym w grupie leczonej okrelizumabem, a całkowita częstość jej występowania wyniosła 34,3% w porównaniu z 9,9% w grupie pacjentów leczonych interferonem beta-1a (wlew placebo). Częstość występowania reakcji związanych z wlewem była najwyższa w czasie podawania dawki 1., we wlewie 1. (27,5%) i malała w miarę upływu czasu do < 10% podczas podawania dawki 4. Większość reakcji związanych z wlewem w obu grupach leczenia miała nasilenie łagodne do umiarkowanego. Odpowiednio 21,7% i 10,1% pacjentów leczonych okrelizumabem doświadczyło łagodnych i umiarkowanych reakcji związanych z wlewem. U 2,4% pacjentów wystąpiły ciężkie reakcje związane z wlewem, a u 0,1% pacjentów reakcje związane z wlewem stanowiły zagrożenie życia. Patrz punkt 4.4.
W badaniu klinicznym kontrolowanym placebo (PPMS) reakcja związana z wlewem była najczęstszymziałaniem niepożądanym w grupie leczonej okrelizumabem, a całkowita częstość jej występowania wyniosła 40,1% w porównaniu z 25,5% w grupie placebo. Częstość występowania reakcji związanych z wlewem była najwyższa w czasie podawania dawki 1., we wlewie 1. (27,4%) i malała wraz z podawaniem kolejnych dawek do < 10% przy dawce 4. Większy odsetek pacjentów w każdej grupie doświadczył reakcji związanych z wlewem w trakcie pierwszego wlewu każdej dawki w porównaniu z drugim wlewem tej samej dawki. Większość reakcji związanych z wlewem miała nasilenie łagodne do umiarkowanego. Odpowiednio 26,7% i 11,9% pacjentów leczonych okrelizumabem doświadczyło łagodnych i umiarkowanych reakcji związanych z wlewem, a u 1,4% pacjentów wystąpiły ciężkie reakcje związane z wlewem. Nie obserwowano reakcji związanych z wlewem zagrażających życiu. Patrz punkt 4.4.
W okresie leczenia kontrolowanego i w fazie OLE badań klinicznych dotyczących RMS i PPMS pacjenci otrzymali około 20 dawek okrelizumabu. Częstość występowania IRR zmniejszyła się do <4% do czasu podania dawki 4. w fazie OLE u pacjentów z RMS oraz do <5% do czasu podania dawki 5. w fazie OLE u pacjentów z PPMS. Wraz z podawaniem kolejnych dawek w fazie OLE częstość występowania IRR pozostała niska. W fazie OLE większość IRR miała nasilenie łagodne.
Alternatywny krótszy wlew kolejnych dawek
W badaniu (dodatkowe badanie z krótszym czasem wlewu MA30143) opracowanym w celu scharakteryzowania profilu bezpieczeństwa krótszych (2-godzinnych) wlewów okrelizumabu u pacjentów z rzutowo-remisyjną postacią stwardnienia rozsianego, częstość występowania, nasilenie i rodzaj objawów w przebiegu reakcji związanych z wlewem były spójne z analogicznymi obserwacjami dla wlewów podawanych przez 3,5 godziny (patrz punkt 5.1). Całkowita liczba koniecznych interwencji była niska w obu grupach otrzymujących wlew, jednak więcej interwencji (zmniejszenie prędkości lub czasowe przerwanie wlewu) w ramach postępowania z IRR było koniecznych w grupie otrzymującej krótszy (2-godzinny) wlew w porównaniu z grupą otrzymującą wlew trwający 3,5 godziny (odpowiednio: 8,7% w porównaniu z 4,8%).
Zakażenie
W badaniach z grupą kontrolną otrzymującą substancję czynną u pacjentów z RMS zakażenia wystąpiły u 58,5% pacjentów otrzymujących okrelizumab w porównaniu z 52,5% pacjentów otrzymujących interferon beta 1a. Ciężkie zakażenia wystąpiły u 1,3% pacjentów przyjmujących okrelizumab w porównaniu z 2,9% pacjentów przyjmujących interferon beta-1a. W badaniu z grupą kontrolną otrzymującą placebo u pacjentów z PPMS zakażenia wystąpiły u 72,2% pacjentów otrzymujących okrelizumab w porównaniu z 69,9% pacjentów otrzymujących placebo.
Poważne zakażenia wystąpiły u 6,2% pacjentów przyjmujących okrelizumab w porównaniu z 6,7% pacjentów przyjmujących placebo.
Wszyscy pacjenci przeszli na okrelizumab podczas fazy OLE w obu badaniach RMS i PPMS. Podczas fazy OLE u pacjentów z RMS i PPMS całkowite ryzyko ciężkich zakażeń nie zwiększyło się w porównaniu z ryzykiem obserwowanym w okresie leczenia kontrolowanego. Jak zauważono w kontrolowanym okresie badań częstość występowania ciężkich zakażeń było obserwowane u pacjentów u pacjentów z PPMS pozostawała większa niż u pacjentów z RMS.
Zgodnie z wcześniejszą analizą czynników ryzyka ciężkich zakażeń w przebiegu schorzeń autoimmunologicznych innych niż SM (patrz punkt 4.4) przeprowadzono wielowymiarową analizę czynników ryzyka ciężkich zakażeń z wykorzystaniem skumulowanych danych z około 10 lat ekspozycji, pochodzących z okresu leczenia kontrolowanego i fazy OLE rejestracyjnych badań klinicznych. Czynniki ryzyka ciężkich zakażeń u pacjentów z RMS obejmują występowanie co najmniej 1 choroby współistniejącej, niedawno przebyty kliniczny rzut choroby oraz EDSS (ang. Expanded Disability Status Scale, rozszerzona skala niesprawności ruchowej) ≥ 6,0. Czynniki ryzyka ciężkich zakażeń u pcjantów z PPMS obejmują wskaźnik masy ciała większy niż 25 kg/m2, występowanie co najmniej 2 chorób współistniejących, EDSS ≥ 6,0 oraz IgM < dolnej granicy normy (DGN). Choroby współistniejące obejmują między innymi schorzenia układu sercowo-naczyniowego, nerek i układu moczowego, wcześniejsze zakażenia i depresję.
Zakażenia układu oddechowego
Odsetek zakażeń układu oddechowego był wyższy wśród pacjentów przyjmujących okrelizumab niż w grupach pacjentów przyjmujących interferon beta-1a i placebo.
W badaniach klinicznych z udziałem pacjentów z RMS zakażenie górnych dróg oddechowych wystąpiło u 39,9% pacjentów przyjmujących produkt leczniczy Ocrevus i u 33,2% pacjentów przyjmujących interferon beta-1a, natomiast zakażenie dolnych dróg oddechowych wystąpiło u 7,5% pacjentów przyjmujących produkt leczniczy Ocrevus i 5,2% pacjentów przyjmujących interferon beta-1a.
W badaniu klinicznym z udziałem pacjentów z PPMS zakażenie górnych dróg oddechowych wystąpiło u 48,8% pacjentów przyjmujących okrelizumab i u 42,7% pacjentów przyjmujących placebo, natomiast zakażenie dolnych dróg oddechowych wystąpiło u 9,9% pacjentów przyjmujących okrelizumab i 9,2% pacjentów przyjmujących placebo.
Zakażenia układu oddechowego zgłaszane u pacjentów leczonych okrelizumabem były głównie łagodne do umiarkowanych (80–90%).
Opryszczka
W badaniach klinicznych z grupą kontrolną otrzymującą substancję czynną (RMS) zakażenia wirusem opryszczki były zgłaszane częściej u pacjentów przyjmujących okrelizumab niż u pacjentów przyjmujących interferon beta-1a i obejmowały one półpasiec (2,1% w porównaniu z 1%), opryszczkę zwykłą (0,7% w porównaniu z 0,1%), opryszczkę jamy ustnej (3% w porównaniu z 2,2%), opryszczkę narządów płciowych (0,1% w porównaniu z 0%) i zakażenie wirusem opryszczki (0,1% w porównaniu z 0%). Wszystkie zakażenia miały nasilenie łagodne do umiarkowanego, oprócz jednego
zdarzenia w stopniu 3., zakażenia ustępowały po zastosowaniu standardowego leczenia.
W badaniu klinicznym z grupą kontrolną otrzymującą placebo (PPMS) wyższy odsetek pacjentów z opryszczką jamy ustnej (2,7% w porównaniu z 0,8%) obserwowano w grupie przyjmującej okrelizumab.
Odchylenia w wynikach badań laboratoryjnych
Immunoglobuliny
Leczenie okrelizumabem prowadziło do spadku całkowitego stężenia immunoglobulin w kontrolowanym okresie rejestracyjnych badań klinicznych, wynikającego głównie ze spadku miana IgM.
Dane z badań klinicznych pochodzące z okresu leczenia kontrolowanego i z fazy OLE rejestracyjnych badań klinicznych wykazały związek pomiędzy zmniejszonym stężeniem IgG (a w mniejszym stopniu także IgM lub IgA) a zwiększoną częstością występowania ciężkich zakażeń. U 2,1% pacjentów z RMS ciężkie zakażenie wystąpiło w okresie, gdy IgG < DGN; u 2,3% pacjentów z PPMS ciężkie zakażenie wystąpiło w okresie, gdy IgG < DGN. Różnica w częstości występowania ciężkich zakażeń pomiędzy pacjentami z IgG < DGN a pacjentami z IgG ≥ DGN nie zwiększyła się w miarę upływu czasu. Rodzaj, nasilenie, latencja, czas trwania i wynik leczenia ciężkich zakażeń obserwowanych podczas epizodów zmniejszenia stężenia immunoglobulin do poziomów poniżej DGN były spójne ze wszystkimi ciężkimi zakażeniami obserwowanymi u pacjentów leczonych okrelizumabem w okresie leczenia kontrolowanego i w fazie OLE. Przez cały 10-letni okres ciągłego leczenia okrelizumabem średnie stężenie IgG u pacjentów z RMS i PPMS pozostawało na poziomie wartości powyżej DGN.
Limfocyty
W RMS zmniejszenie liczby limfocytów < DGN było obserwowane u 20,7% pacjentów otrzymujących okrelizumab w porównaniu z 32,6% pacjentów leczonych interferonem beta-1a. W PPMS zmniejszenie liczby limfocytów < DGN było obserwowane u 26,3% pacjentów leczonych okrelizumabem w porównaniu z 11,7% pacjentów otrzymujących placebo.
Większość tych spadków zgłaszanych u pacjentów leczonych okrelizumabem miała nasilenie stopnia 1. (< DGN – 800 komórek/mm3) i 2. (pomiędzy 500 a 800 komórek/mm3). U około 1% pacjentów z grupy otrzymującej okrelizumab wystąpiła limfopenia stopnia 3. (pomiędzy 200 a 500 komórek/mm3). U żadnego pacjenta nie zgłoszono limfopenii stopnia 4. (< 200 komórek/mm3).
Podczas epizodów potwierdzonego spadku całkowitej liczby limfocytów u pacjentów leczonych okrelizumabem obserwowano zwiększoną częstość występowania ciężkich zakażeń. Liczba ciężkich zakażeń była zbyt mała, by wyciągnąć jednoznaczne wnioski.
Neutrofile
W okresie leczenia kontrolowanego substancją czynną (RMS) spadek liczby neutrofilów < DGN obserwowano u 14,7% pacjentów leczonych okrelizumabem w porównaniu z 40,9% pacjentów leczonych interferonem beta-1a. W badaniu klinicznym z grupą kontrolną otrzymującą placebo (PPMS) odsetek pacjentów ze zmniejszoną liczbą neutrofilów był wyższy w grupie otrzymującej okrelizumab (12,9%) niż w grupie przyjmującej placebo (10,0%); wśród tych osób neutropenia stopnia 2. lub wyższego występowała u większego odsetka pacjentów (4,3%) z grupy otrzymującej okrelizumab w porównaniu z 1,3% w grupie placebo; u około 1% pacjentów z grupy otrzymującej okrelizumab wystąpiła neutropenia stopnia 4. w porównaniu z 0% w grupie otrzymującej placebo.
Większość przypadków zmniejszenia liczby neutrofilów była przemijająca (zdarzenie obserwowane jednorazowo u danego pacjenta leczonego okrelizumabem) i miała nasilenie stopnia 1. (pomiędzy < DGN a 1500 komórek/mm3) i 2. (pomiędzy 1000 a 1500 komórek/mm3). Ogółem, neutropenia stopnia 3. lub 4. wystąpiła u około 1% pacjentów z grupy leczonej okrelizumabem. Jeden pacjent z neutropenią stopnia 3. (pomiędzy 500 a 1000 komórek/mm3) i jeden pacjent z neutropenią stopnia 4. (< 500 komórek/mm3) wymagał specjalnego leczenia czynnikiem stymulującym wzrost kolonii granulocytów; pacjenci ci kontynuowali leczenie okrelizumabem po tym epizodzie. Neutropenia może wystąpić kilka miesięcy po podaniu okrelizumabu (patrz punkt 4.4).
Inne
Jeden pacjent, który otrzymał okrelizumab w dawce 2000 mg, zmarł z powodu zespołu ogólnoustrojowej reakcji zapalnej (ang. systemic inflammatory response syndrome, SIRS) o nieznanej etiologii po wykonaniu badania obrazowego metodą rezonansu magnetycznego (MRI) 12 tygodni po ostatnim wlewie; do wystąpienia SIRS mogła przyczynić się reakcja rzekomoanafilaktyczna na środek kontrastowy z gadolinem stosowany w MRI.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
Istnieje ograniczone doświadczenie z badań klinicznych dotyczące podania dawek wyższych niż zatwierdzona dawka okrelizumabu. Do chwili obecnej najwyższa dawka, jaką badano u pacjentów ze stwardnieniem rozsianym wynosi 2000 mg i została podana w dwóch wlewach dożylnych po 1000 mg w odstępie 2 tygodni (badanie fazy II nad ustaleniem optymalnej dawki w leczeniu RRMS) i dawka 1200 mg podana we wstrzyknięciu podskórnym (badanie fazy Ib nad ustaleniem optymalnej dawki). Działania niepożądane były spójne z profilem bezpieczeństwa stosowania opisanym w rejestracyjnych badaniach klinicznych.
Brak jest swoistego antidotum w przypadku przedawkowania; należy natychmiast przerwać wlew i obserwować pacjenta pod kątem reakcji związanych z wlewem (patrz punkt 4.4).
Grupa farmakoterapeutyczna: Leki immunosupresyjne, selektywne leki grupa przeciwciała monoklonalne, kod ATC: L04AG08
Mechanizm działania
Okrelizumab jest rekombinowanym humanizowanym przeciwciałem monoklonalnym, które jest selektywnie skierowane przeciwko limfocytom B z ekspresją antygenu CD20.
CD20 jest powierzchniowym antygenem znajdującym się na limfocytach pre-B, dojrzałych limfocytach B i limfocytach B pamięci, który nie podlega ekspresji na limfoidalnych komórkach macierzystych i komórkach plazmatycznych.
Dokładny mechanizm odpowiedzialny za terapeutyczne działanie okrelizumabu w stwardnieniu rozsianym nie jest w pełni wyjaśniony, ale zakłada się, że obejmuje on immunomodulację poprzez zmniejszenie liczby i pogorszenie funkcjonowania limfocytów B z ekspresją antygenu CD20. Po połączeniu się z powierzchnią komórki, okrelizumab wybiórczo usuwa limfocyty B z ekspresją antygenu CD20 w mechanizmie fagocytozy komórkowej zależnej od przeciwciał (ang. antibody- dependent cellular phagocytosis, ADCP), cytotoksyczności komórkowej zależnej od przeciwciał (ang. antibody-dependent cellular cytotoxicity, ADCC), cytotoksyczności zależnej od dopełniacza (ang. complement-dependent cytotoxicity, CDC) i apoptozy. Zdolność odnowy limfocytów B i wcześniejsza odporność humoralna zostają zachowane. Ponadto, odporność wrodzona i całkowita liczba limfocytów T nie są zmienione.
Działanie farmakodynamiczne
Leczenie okrelizumabem prowadzi do szybkiej deplecji limfocytów B CD19+ we krwi w ciągu 14 dni po leczeniu (pierwszy punkt czasowy oceny), co stanowi oczekiwane działanie farmakologiczne. Działanie takie utrzymywało się przez cały okres leczenia. Do ustalenia liczby limfocytów B wykorzystuje się CD19, ponieważ obecność okrelizumabu zaburza rozpoznawanie CD20 w badaniu.
W badaniach III fazy, pomiędzy poszczególnymi dawkami okrelizumabu odnowę limfocytów B (> DGN lub wartości wyjściowych) obserwowano u nie więcej niż 5% pacjentów w przynajmniej jednym punkcie czasowym. Zakres i czas trwania deplecji limfocytów B był spójny w badaniach z udziałem pacjentów z PPMS i RMS.
Najdłuższy okres obserwacji po ostatnim podaniu wlewu (badanie II fazy WA21493, n=51) wskazuje, że mediana czasu do odnowy limfocytów B (powrót do wartości wyjściowych / DGN, w zależności od tego, co nastąpiło wcześniej) wyniosła 72 tygodnie (zakres 27 - 175 tygodni). U 90% wszystkich pacjentów powrót liczby limfocytów B do DGN lub do wartości początkowych nastąpił w ciągu około dwóch i pół roku od podania ostatniego wlewu.
Skuteczność kliniczna i bezpieczeństwo stosowania
Rzutowe postacie stwardnienia rozsianego (RMS)
Skuteczność i bezpieczeństwo stosowania okrelizumabu oceniano w dwóch randomizowanych, podwójnie zaślepionych, podwójnie maskowanych badaniach klinicznych z grupą kontrolną otrzymującą substancję czynną (WA21092 i WA21093), prowadzonych według takiego samego planu, z udziałem pacjentów z rzutowymi postaciami stwardnienia rozsianego (według kryteriów McDonalda z 2010 r.) z potwierdzoną aktywnością choroby (definiowaną na podstawie cech klinicznych lub radiologicznych) w ciągu ostatnich dwóch lat. Plan badania i wyjściową charakterystykę badanej populacji przedstawiono w Tabeli 3.
Dane demograficzne i wyjściowa charakterystyka badanej populacji były dobrze wyważone w obu grupach leczenia. Pacjenci przyjmujący okrelizumab (Grupa A) otrzymywali dawkę 600 mg co 6 miesięcy (Dawka 1.: 2 wlewy dożylne po 300 mg, podane w odstępie dwóch tygodni, kolejne dawki podano w pojedynczym wlewie dożylnym 600 mg). Pacjentom w Grupie B podawano interferon beta-1a w dawce 44 µg we wstrzyknięciach podskórnych 3 razy w tygodniu.
Tabela 3: Plan badania, charakterystyka demograficzna i wyjściowa pacjentów.
|
|
Badanie 1 |
Badanie 2 |
||
|
Nazwa badania |
WA21092 (OPERA I)(n=821) |
WA21093 (OPERA II)(n=835) |
||
|
Plan badania |
||||
|
Populacja badania |
Pacjenci z rzutowymi postaciami stwardnienia rozsianego |
|||
|
Historia choroby w fazie przesiewowej |
Przynajmniej dwa rzuty w ciągu ostatnich dwóch lat lub jeden rzut w minionym roku; wynik EDSS* pomiędzy 0 a 5,5włącznie |
|||
|
Czas trwania badania |
2 lata |
|||
|
Grupy leczenia |
Grupa A: Okrelizumab 600 mg Grupa B: interferon beta-1a 44 µg podskórnie (IFN) |
|||
|
Charakterystyka wyjściowa |
Okrelizumab |
IFN |
Okrelizumab |
IFN |
|
Średni wiek (lata) |
37,1 |
36,9 |
37,2 |
37,4 |
|
Zakres wieku (lata) w chwili włączenia do badania |
18 - 56 |
18 - 55 |
18 - 55 |
18 - 55 |
|
Rozkład płci (% mężczyzn/% kobiet) |
34,1/65,9 |
33,8/66,2 |
35,0/65,0 |
33,0/67,0 |
|
Średni czas/Mediana czasutrwania choroby od rozpoznania (lata) |
3,82/1,53 |
3,71/1,57 |
4,15/2,10 |
4,13/1,84 |
|
Pacjenci nieleczeniwcześniej lekamimodyfikującymi przebiegchoroby (%)** |
73,4 |
71,0 |
72,7 |
74,9 |
|
Średnia liczba rzutów w ostatnim roku |
1,31 |
1,33 |
1,32 |
1,34 |
|
Odsetek pacjentów zezmianami w obrazach T1-zależnych wzmacniającymi się po gadolinie |
42,5 |
38,1 |
39,0 |
41,4 |
|
Średni wynik w skali EDSS* |
2,82 |
2,71 |
2,73 |
2,79 |
* ang. Expanded Disability Status Scale, rozszerzona skala niesprawności ruchowej
** Pacjenci nieleczeni lekiem modyfikującym przebieg choroby (ang. disease-modifying therapy, DMT) w okresie 2 lat przed randomizacją.
W Tabeli 4 i na Rycinie 1 przedstawiono najważniejsze wyniki dotyczące skuteczności klinicznej i wyniki dotyczące skuteczności widoczne w badaniu rezonansem magnetycznym.
Wyniki omówionych badań wykazują, że okrelizumab istotnie hamuje rzuty choroby, zmniejsza subkliniczną aktywność choroby ocenianą w badaniu rezonansem magnetycznym oraz hamuje postęp choroby w porównaniu z interferonem beta-1a podawanym podskórnie w dawce 44 µg.
Tabela 4: Najważniejsze kliniczne punkty końcowe i punkty końcowe oceniane w MRI w badaniach WA21092 i WA21093 (RMS)
|
Punkty końcowe |
Badanie 1: WA21092 (OPERA I) |
Badanie 2: WA21093 (OPERA II) |
||
|
Okrelizumab |
IFN |
Okrelizumab |
IFN |
|
|
Kliniczne punkty końcowe |
|
|||
|
Roczny wskaźnik rzutów (ARR) (pierwszorzędowy punktkońcowy) |
0,156 |
0,292 |
0,155 |
0,290 |
|
Względna redukcja |
46 % (p<0,0001) |
47 % (p<0,0001) |
||
|
Odsetek pacjentów z 12-tygodniową potwierdzoną progresją niesprawności3 Redukcja ryzyka (analiza zbiorcza1)Redukcja ryzyka (poszczególne badania2) |
9,8% Okrelizumab w porównaniu z 15,2% IFN 40% (p=0,0006)7 |
|||
|
43 % (p=0,0139)7 |
37 % (p=0,0169)7 |
|||
|
Odsetek pacjentów z 24-tygodniową potwierdzoną progresją niesprawności (CDP)3 Redukcja ryzyka (analiza zbiorcza1) Redukcja ryzyka (poszczególne badania2) |
7,6% Okrelizumab w porównaniu z 12,0% IFN |
|||
|
43 % (p=0,0278)7 |
37 % (p=0,0370)7 |
|||
|
Odsetek pacjentów z przynajmniej 12-tygodniową potwierdzoną poprawą w zakresie niesprawności4 |
20,7% Okrelizumab w porównaniu z 15,6% IFN |
|||
|
Względny wzrost (analiza zbiorcza1) Względny wzrost (poszczególne badania2) |
33% (p=0,0194) |
|||
|
61% (p=0,0106) |
14% (p=0,4019) |
|||
|
Odsetek pacjentów bez rzutów choroby po 96 tygodniach2 |
80,4% |
66,7% |
78,9% |
64,3% |
|
(p<0,0001) |
(p<0,0001) |
|||
|
Odsetek pacjentów z brakiem aktywności choroby |
48% |
29% |
48% |
25% |
|
Względny wzrost2 |
64% (p<0,0001) |
89% (p<0,0001) |
||
|
Punkty końcowe mierzone za pomocą MRI |
|
|||
|
Średnia liczba zmian w obrazach T1 zależnych wzmacniających się po gadolinie na jedno badanie MRI |
0,016 |
0,286 |
0,021 |
0,416 |
|
Względna redukcja |
94% (p<0,0001) |
95% (p<0,0001) |
||
|
Średnia liczba nowych i (lub) powiększających się zmian hiperintensywnych w obrazach T2-zależnych na jedno |
0,323 |
1,413 |
0,325 |
1,904 |
|
Względna redukcja |
77% (p<0,0001) |
83% (p<0,0001) |
||
|
Zmiana procentowa objętości mózgu od tygodnia 24. do tygodnia 96. |
-0,572 |
-0,741 |
-0,638 |
-0,750 |
|
Względna redukcja utraty objętości mózgu |
22,8% (p=0,0042)6 |
14,9% (p=0,0900) |
||
1 Dane zebrane prospektywnie z Badania 1 i 2
2 Niekonfirmacyjna analiza wartości p; nie jest częścią predefiniowanej hierarchii testowania
3 CDP definiowana jako wzrost o ≥ 1,0 punkt w stosunku do wyjściowego wyniku w skali EDSS dla pacjentów z wynikiem wyjściowym 5,5 lub niższym, lub ≥ 0,5, jeżeli wynik wyjściowy jest wyższy niż 5,5, estymatory Kaplana- Meiera w tygodniu 96.
4 Definiowany jako spadek o ≥ 1,0 punkt w stosunku do wyjściowego wyniku w skali EDSS dla pacjentów z wyjściowym wynikiem w skali EDSS ≥ 2 i ≤ 5,5 lub ≥ 0,5, gdy wynik wyjściowy wynosi > 5,5. Pacjenci z wynikiem wyjściowym < 2 nie zostali uwzględnieni w analizie.
5 NEDA definiowany jako brak rzutów zgodnych z definicją w protokole, 12-tygodniowej CDP i wszelkiej aktywność widocznej w badaniu MRI (zmiany w obrazach T1-zależnych podlegających wzmocnieniu po gadolinie bądź nowe lub powiększające się zmiany w obrazach T2-zależnych) w całym 96- tygodniowym okresie leczenia. Wynik analizy eksploracyjnej obejmuje całą populację zgodną z intencją leczenia (ang. intent-to-treat, ITT).
6 Niekonfirmacyjna wartość p; procedura hierarchicznego testowania zakończona przed osiągnięciem punktu końcowego.
7 Log-rank test
8Potwierdzone rzuty (którymtowarzyszyła klinicznie istotnazmiana w EDSS)
Rycina 1: Wykres Kaplana-Meiera przedstawiający czas do początku potwierdzonej progresji niesprawności utrzymującej się przez przynajmniej 12 tygodni, gdy początkowe zdarzenie pogorszenia stanu neurologicznego miało miejsce w trakcie okresu leczenia metodą podwójnie ślepej próby (zbiorcza populacja ITT w badaniach WA21092 i WA21093)*
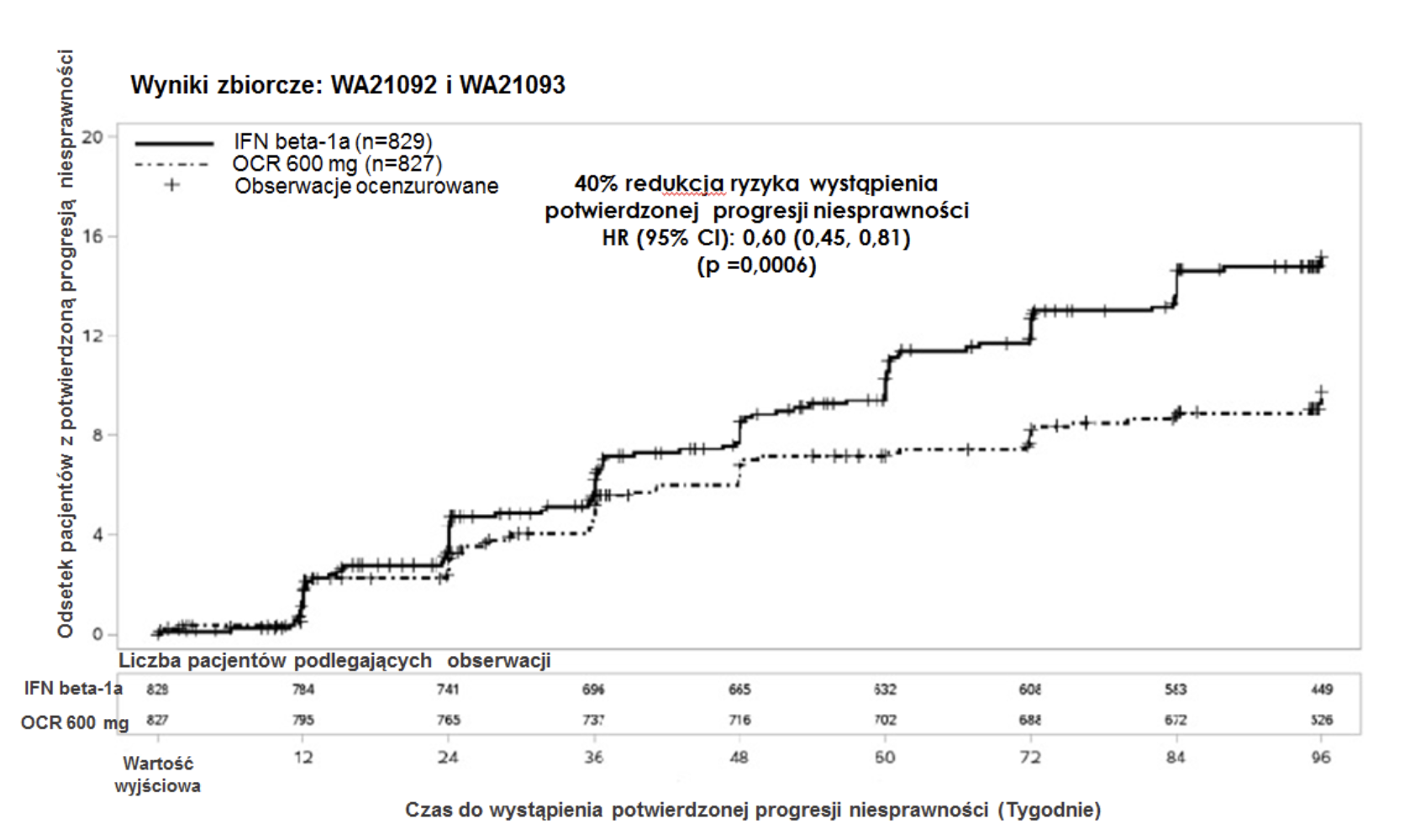
*Predefiniowana analiza zbiorcza badań WA21092 i WA21093
Wyniki predefiniowanych analiz zbiorczych czasu do wystąpienia potwierdzonej progresji niesprawności utrzymującej się przez przynajmniej 12 tygodni (40% redukcja ryzyka w grupie leczonej okrelizumabem w porównaniu z interferonem beta-1a (p=0,0006) były w wysokim stopniu spójne z wynikami utrzymującymi się przez przynajmniej 24 tygodnie (40% redukcja ryzyka w grupie leczonej okrelizumabem w porównaniu z interferonem beta- 1a, p=0,0025).
Do badań włączano pacjentów z aktywną chorobą. Do populacji badanej należeli zarówno pacjenci nieotrzymujący wcześniej aktywnego leczenia, jak i pacjenci wcześniej leczeni z niewystarczającą odpowiedzią na leczenie, co definiowano na podstawie cech klinicznych lub radiologicznych. Analiza populacji pacjentów różniących się wyjściową aktywnością choroby, w tym pacjentów z aktywną i wysoce aktywną chorobą wykazała, że skuteczność okrelizumabu w odniesieniu do rocznego wskaźnika rzutów i wskaźnika 12-tygodniowej potwierdzonej progresji niesprawności była spójna z wynikami dla populacji całkowitej.
Pierwotnie postępująca postać stwardnienia rozsianego (PPMS)
Skuteczność i bezpieczeństwo stosowania okrelizumabu oceniano również w randomizowanym, podwójnie zaślepionym badaniu klinicznym z grupą kontrolną otrzymującą placebo z udziałem pacjentów z pierwotnie postępującą postacią stwardnienia rozsianego (badanie WA25046), którzy znajdowali się na wczesnym etapie przebiegu choroby według głównych kryteriów włączenia, tj. wiek od 18 do 55 lat włącznie; EDSS w chwili przesiewu od 3,0 do 6,5 punktu; czas trwania od pierwszych objawów stwardnienia rozsianego mniej niż 10 lat u pacjentów z wynikiem EDSS w chwili przesiewu 5,0 lub mniej niż 15 lat u pacjentów z wynikiem EDSS w chwili przesiewu >5,0. W odniesieniu do aktywności choroby, cechy charakterystyczne dla aktywności zapalnej, nawet w postępującej postaci stwardnienia rozsianego, mogą być związane z cechami radiologicznymi (tj. zmiany w obrazach T1-zależnych wzmacniających się po gadolinie i (lub) aktywne [nowe lub powiększające się] zmiany T2. Dane uzyskane z MRI należy wykorzystać do potwierdzenia aktywności zapalnej u wszystkich pacjentów. Pacjenci w wieku ponad 55 lat nie byli badani. Plan badania i charakterystykę wyjściową badanej populacji przedstawiono w Tabeli 5.
Dane demograficzne i wyjściowa charakterystyka były dobrze wyważone w obu grupach leczenia. MRI głowy wykazywało cechy charakterystyczne dla aktywności zapalnej poprzez zmiany w obrazach T1-zależnych wzmacniających się po gadolinie lub zmiany T2.
W trakcie badania fazy 3 z udziałem pacjentów z PPMS pacjentom podawano dawkę 600 mg okrelizumabu co 6 miesięcy w dwóch wlewach po 300 mg w odstępie dwóch tygodni, przez cały czas trwania leczenia. Wlewy dawki 600 mg podawane pacjentom z RMS i wlewy dawki 2 x 300 mg w PPMS wykazywały spójne profile farmakokinetyczne/farmakodynamiczne. Profile reakcji związanej z wlewem dla każdego podania były również podobne, niezależnie od tego, czy dawka 600 mg była podawana jako pojedynczy wlew 600 mg czy jako dwa wlewy po 300 mg w odstępie dwóch tygodni (patrz punkt 4.8 i 5.2), ale z powodu ogólnie większej liczby podań wlewów w schemacie dawkowania 2 x 300 mg, całkowita liczba reakcji związanych z wlewem była wyższa. Dlatego rekomenduje się, aby po podaniu dawki 1. okrelizumabu podawać w pojedynczym wlewie 600 mg (patrz punkt 4.2), aby zmniejszyć całkowitą liczbę podań wlewów (z równoczesnym profilaktycznym podaniem metyloprednizolonu i leku antyhistaminowego) i reakcji związanych z wlewem.
Tabela 5: Plan badania, dane demograficzne i wyjściowa charakterystyka badania WA25046
|
Nazwa badania |
Badanie WA25046 ORATORIO (n=732) |
|
|
Plan badania |
||
|
Badana populacja |
Pacjenci z pierwotnie postępującą postacią stwardnienia rozsianego |
|
|
Czas trwania badania |
Zależny od zdarzeń (Minimum 120 tygodni i 253 zdarzenia potwierdzonej progresji niesprawności)(Mediana czasu obserwacji: Okrelizumab 3,0 lata, Placebo 2,8 lat) |
|
|
Przebieg choroby w fazie przesiewowej |
Wiek 18-55 lat, wynik EDSS 3,0 do 6,5 |
|
|
Grupy leczenia |
Grupa A: Okrelizumab 600 mg |
|
|
Charakterystyka wyjściowa |
Okrelizumab 600 mg (n=488) |
Placebo (n=244) |
|
Średni wiek (lata) |
44,7 |
44,4 |
|
Zakres wieku (lata) wmomencie włączenia do badania |
20 – 56 |
18 – 56 |
|
Rozkład płci (% mężczyzn/% kobiet) |
51,4/48,6 |
49,2/50,8 |
|
Średni czas/Mediana czasu trwania choroby odrozpoznania PPMS (lata) |
2,9/1,6 |
2,8/1,3 |
|
Średni wynik EDSS |
4,7 |
4,7 |
Najważniejsze wyniki skuteczności klinicznej i wyniki dotyczące skuteczności obserwowane w badaniu MRI przedstawiono w Tabeli 6 i na Rycinie 2.
Wyniki badania pokazują, że okrelizumab istotnie opóźnia progresję choroby i zmniejsza pogorszanie szybkości chodzenia w porównaniu z placebo.
Tabela 6: Najważniejsze kliniczne punkty końcowe i punkty końcowe mierzone za pomocą
rezonansu magnetycznego w badaniu WA25046 (PPMS)
|
|
Badanie 3 |
|
|
Punkty końcowe |
WA25046 (Oratorio) |
|
|
Okrelizumab 600mg (n=488) |
Placebo |
|
|
Kliniczne punkty końcowe |
||
|
Pierwszorzędowy punkty końcowy dotyczący skuteczności Odsetek pacjentów z 12-tygodniową potwierdzoną progresją niesprawności1 (pierwszorzędowy punkt końcowy) Redukcja ryzyka |
30,2% |
34,0% |
|
24%(p=0,0321) |
||
|
Odsetek pacjentów z 24-tygodniową potwierdzoną progresją niesprawności1 |
28,3% |
32,7% |
|
Redukcja ryzyka |
25%(p=0,0365) |
|
|
Zmiana procentowa w teście szybkości chodu naodcinku 7,5 m od wartości wyjściowej do tygodnia 120. |
38,9 |
55,1 |
|
Względna redukcja szybkości pogorszenia czasu chodzenia |
29,4%(p=0,0404) |
|
|
Punkty końcowe mierzone za pomocą rezonansu magnetycznego |
||
|
Procentowa zmiana objętości zmian hiperintensywnych w obrazach T2-zależnych, od wartości wyjściowej do tygodnia 120. |
-3,4 |
7,4 |
|
(p<0,0001) |
||
|
Procentowa zmiana objętości mózgu od tygodnia 24. do tygodnia 120. |
-0,902 |
-1,093 |
|
Względna redukcja szybkości utraty objętości mózgu |
17,5%(p=0,0206) |
|
1 Definiowany jako wzrost o ≥ 1,0 punkt od wyjściowego wyniku w skali EDSS u pacjentów z wynikiem wyjściowym wynoszącym 5,5 lub mniej, lub o ≥ 0,5, jeżeli wynik wyjściowy jest większy niż 5.5, estymatory Kaplana-Meiera w tygodniu 120.
Rycina 2: Wykres Kaplana-Meiera przedstawiający czas do początku potwierdzonej progresji niesprawności utrzymującego się przez co najmniej 12 tygodni, gdy początkowe zdarzenie pogorszenia stanu neurologicznego miało miejsce w trakcie podwójnie zaślepionego okresu leczenia (populacja ITT w badaniu WA25046)*
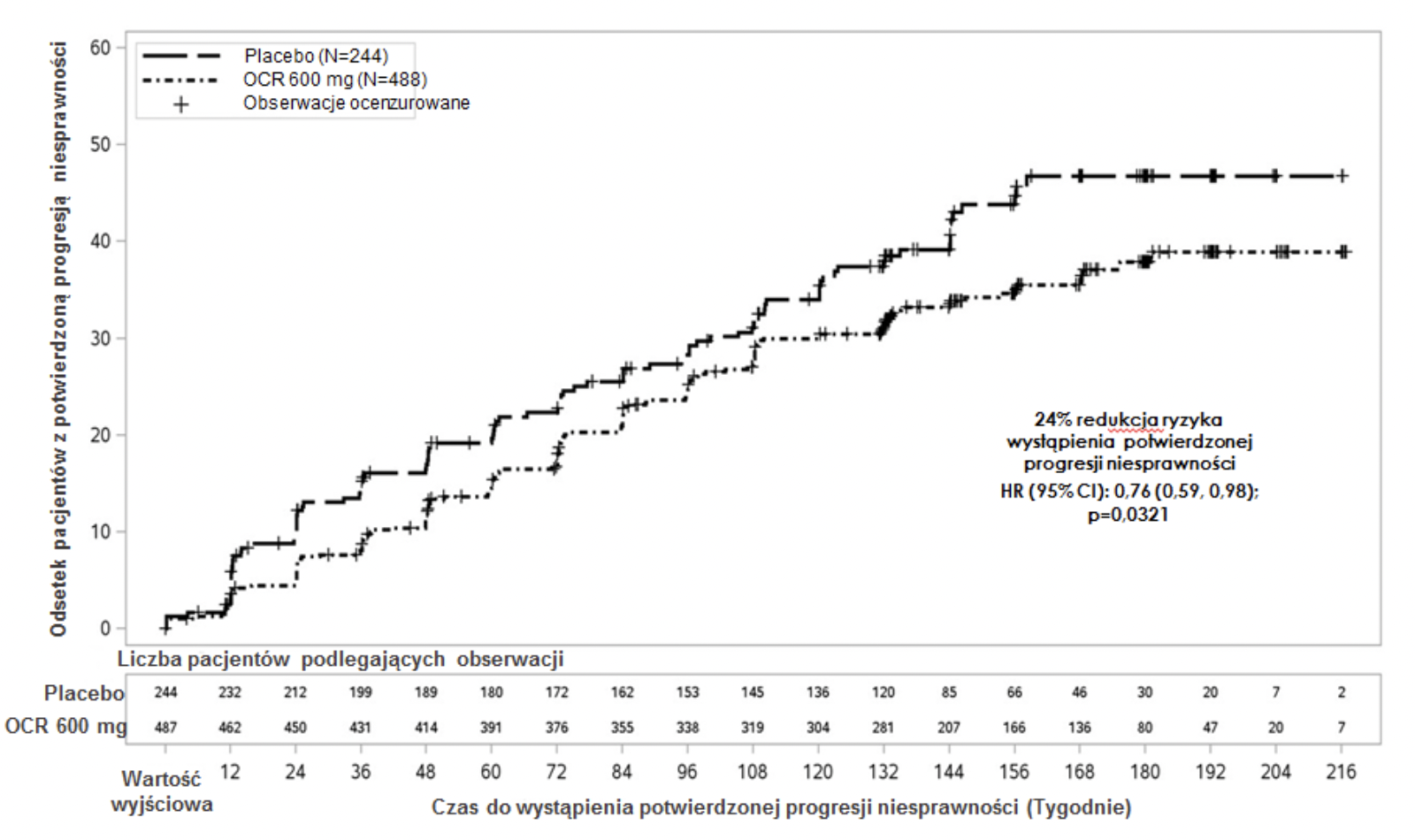
* U wszystkich pacjentów w tej analizie okres obserwacji trwał przynajmniej 120 tygodni. Analiza pierwotna została przeprowadzona w oparciu o wszystkie odnotowane zdarzenia.
Analiza predefiniowanych podgrup o nieokreślonej mocy w odniesieniu do pierwszorzędowego punktu końcowego sugeruje, że młodsi pacjenci lub ci ze zmianami w obrazach T1-zależnych wzmacniających się po gadolinie odnoszą większą korzyść z leczenia niż pacjenci, którzy są starsi i nie mają zmian w obrazach T1-zależnych wzmacniających się po gadolinie (≤ 45 lat: HR 0,64 [0,45; 0,92], >45 lat: HR 0,88 [0,62; 1,26]; pacjenci ze zmianami w obrazach T1-zależnych wzmacniających się po gadolinie w punkcie wyjścia: HR 0,65 [0,40–1,06], pacjenci bez zmian w obrazach T1-zależnych wzmacniających się po gadolinie w punkcie wyjścia: HR 0,84 [0,62–1,13]).
Ponadto, analizy post-hoc sugerują, że młodsi pacjenci ze zmianami w obrazach T1-zależnych wzmacniających się po gadolinie w punkcie wyjścia uzyskują lepsze skutki leczenia (≤ 45 lat: HR 0,52 [0,27–1,00]; ≤ 46 lat (mediana wieku w badaniu WA25046); HR 0,48 [0,25–0,92]; <51 lat: HR 0,53 [0,31–0,89]).
Analizy post-hoc przeprowadzono w wydłużonym okresie badania z grupą kontrolną (ang. extended controlled period, ECP), obejmującym leczenie metodą podwójnie ślepej próby i około 9 dodatkowych miesięcy obserwacji z grupą kontrolną, po których następowała kontynuacja udziału w otwartej fazie przedłużonej badania (ang. open-label extension, OLE) lub odstawienie badanego leczenia. Odsetek pacjentów z 24-tygodniową potwierdzoną progresją niesprawności z wynikiem w skali EDSS ≥7,0 (24W-CDP w skali EDSS ≥7,0, czas do poruszania się na wózku inwalidzkim) wyniósł 9,1% w grupie placebo w porównaniu z 4,8% w grupie otrzymującej okrelizumab w tygodniu 144, co spowodowało 47% redukcję ryzyka w odniesieniu do czasu do poruszania się na wózku inwalidzkim (HR 0,53, [0,31; 0,92]) w okresie ECP. Ponieważ wyniki te mają charakter eksploracyjny i obejmują dane po odślepieniu, należy je interpretować z zachowaniem ostrożności.
Dodatkowe badanie z krótszym czasem wlewu
Bezpieczeństwo stosowania krótszego (2-godzinnego) wlewu okrelizumabu oceniano w prospektywnym, wieloośrodkowym, randomizowanym badaniu z grupą kontrolną, prowadzonym w grupach równoległych metodą podwójnie ślepej próby, które było badaniem dodatkowym do badania MA30143 (Ensemble) z udziałem pacjentów z rzutowo-remisyjną postacią stwardnienia rozsianego, nieotrzymujących wcześniej innych terapii modyfikujących przebieg choroby. Pierwszą dawkę okrelizumabu podano w postaci dwóch wlewów po 300 mg (łącznie 600 mg) w odstępie 14 dni. Nie wcześniej niż od drugiej dawki (dawki 2 do 6) pacjenci zostali losowo przydzieleni w stosunku 1:1 do grupy otrzymującej konwencjonalny wlew okrelizumabu trwający około 3,5 godziny co 24 tygodnie lub do grupy otrzymującej krótszy wlew okrelizumabu trwający około 2 godzin co 24 tygodnie. Randomizację poddano stratyfikacji ze względu na region oraz dawkę, przy której pacjenci zostali po raz pierwszy przydzieleni losowo.
Pierwszorzędowym punktem końcowym był odsetek pacjentów z reakcjami związanymi z wlewem, które wystąpiły podczas pierwszego po randomizacji wlewu lub w ciągu 24 godzin po jego zakończeniu. Analizę pierwszorzędową przeprowadzono po randomizacji 580 pacjentów. Odsetek pacjentów z reakcjami związanymi z wlewem, które wystąpiły podczas pierwszego po randomizacji wlewu lub w ciągu 24 godzin po jego zakończeniu wyniósł 24,6% w grupie wlewu o krótszym czasie trwania i 23,1% w grupie wlewu o konwencjonalnym czasie trwania. Różnica między grupami przy stratyfikacji była podobna. Ogółem, dla wszystkich dawek, przy których nastąpiła randomizacja, większość reakcji związanych z wlewem miała nasilenie łagodne lub umiarkowane, a tylko dwie reakcje związane z wlewem miały nasilenie ciężkie – po jednej ciężkiej reakcji związanej z wlewem w każdej z grup. Nie odnotowano zagrażających życiu, śmiertelnych lub ciężkich reakcji związanych z wlewem.
Immunogenność
Pacjenci ze stwardnieniem rozsianym (badania WA21092, WA21093 i WA25046) byli badani w wielu punktach czasowych (przed rozpoczęciem leczenia i co 6 miesięcy po zakończeniu leczenia przez cały czas trwania badania) na obecność przeciwciał przeciwlekowych (ang. anti-drug antibodies, ADA). U 12 spośród 1311 (~1%) pacjentów leczonych okrelizumabem odnotowano dodatni wynik obecności przeciwciał przeciwlekowych występujących podczas leczenia, z czego u dwóch pacjentów badania wykazały obecność przeciwciał neutralizujących. Nie można ocenić wpływu przeciwciał przeciwlekowych występujących podczas leczenia na bezpieczeństwo stosowania i skuteczność z powodu małej częstości występowania przeciwciał przeciwlekowych związanych ze stosowaniem okrelizumabu.
Szczepienia
W randomizowanym, otwartym badaniu z udziałem pacjentów z RMS (n=102), odsetek pacjentów z pozytywną odpowiedzią na szczepionkę przeciwko tężcowi po 8 tygodniach od szczepienia wyniósł 23,9% w grupie okrelizumabu w porównaniu z 54,5% w grupie kontrolnej (nieotrzymującej terapii modyfikującej przebieg choroby, z wyjątkiem interferonu beta). Średnia geometryczna mian przeciwciał przeciwko toksoidowi tężcowemu po 8 tygodniach wyniosła odpowiednio 3,74 i 9,81 IU/ml. Pozytywna odpowiedź na ≥5 serotypów w 23-PPV po 4 tygodniach od szczepienia wyniosła 71,6% w grupie okrelizumabu i 100% w grupie kontrolnej. U pacjentów leczonych okrelizumabem szczepionka przypominająca (13-PCV) podana 4 tygodnie po 23-PPV nie zwiększyła znacząco odpowiedzi na 12 serotypów wspólnych z 23-PPV. Odsetek pacjentów z seroprotekcyjnymi mianami przeciwciał przeciwko pięciu szczepom grypy wahał się w zakresie od 20,0-60,0% i 16,7- 43,8% przed szczepieniem, a 4 tygodnie po szczepieniu odpowiednio od 55,6-80,0% u pacjentów leczonych okrelizumabem i 75,0-97,0% w grupie kontrolnej. Patrz punkt 4.4 i 4.5.
Dzieci i młodzież
Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego Ocrevus w jednej lub kilku podgrupach populacji dzieci i młodzieży w leczeniu stwardnienia rozsianego. Stosowanie u dzieci i młodzieży, patrz punkt 4.2.
Farmakokinetykę okrelizumabu w badaniach nad stwardnieniem rozsianym opisano za pomocą modelu dwukompartmentowego, z klirensem zależnym od czasu i parametrami farmakokinetycznymi typowymi dla przeciwciała monoklonalnego IgG1. Ekspozycja całkowita (AUC w 24-tygodniowym przedziale dawkowania) była identyczna w przypadku dawki 2 x 300 mg stosowanej w PPMS i dawki 1 x 600 mg w badaniach z RMS, co było spodziewane biorąc pod uwagę fakt, że podano taką samą dawkę. Pole pod krzywą (ang. area under the curve, AUCτ) po podaniu czwartej dawki 600 mg okrelizumabu wyniosło 3510 µg/ml•doba, a średnie stężenie maksymalne (ang. maximum concentration, Cmax) wyniosło 212 µg/ml w RMS (wlew 600 mg) i 141 µg/ml w PPMS (wlewy 300 mg).
Wchłanianie
Okrelizumab jest podawany we wlewie dożylnym.
Dystrybucja
Na podstawie farmakokinetyki populacyjnej, szacowana objętość dystrybucji kompartmentu centralnego wyniosła 2,78 l. Objętość dystrybucji kompartmentu obwodowego i klirens między kompartmentami oszacowano na 2,68 l i 0,294 l/dobę.
Metabolizm
Nie przeprowadzono bezpośrednich badań metabolizmu okrelizumabu, ponieważ przeciwciała są usuwane głównie na drodze przemian katabolicznych (tj. rozpadu na peptydy i aminokwasy).
Eliminacja
Stały klirens oszacowano na 0,17 l/dobę, a początkowy klirens zależny od czasu na 0,0489 l/dobę i zmniejszał się on z okresem półtrwania wynoszącym 33 tygodnie. Okres półtrwania w końcowej fazie eliminacji okrelizumabu wyniósł 26 dni.
Szczególne populacje pacjentów
Populacja pediatryczna
Nie przeprowadzono badań farmakokinetyki okrelizumabu u dzieci i młodzieży w wieku poniżej 18 lat.
Pacjenci w podeszłym wieku
Nie przeprowadzono specjalnych badań właściwości farmakokinetycznych okrelizumabu u pacjentów w wieku 55 lat z uwagi na ograniczone doświadczenie kliniczne (patrz punkt 4.2).
Zaburzenia czynności nerek
Nie przeprowadzono formalnych badań właściwości farmakokinetycznych. Pacjenci z łagodnymi zaburzeniami czynności nerek byli włączani do badań klinicznych i nie obserwowano zmian w farmakokinetyce okrelizumabu u tych pacjentów. Brak jest dostępnych danych dotyczących właściwości farmakokinetycznych u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności nerek.
Zaburzenia czynności wątroby
Nie przeprowadzono formalnych badań właściwości farmakokinetycznych. Pacjenci z łagodnymi zaburzeniami czynności wątroby byli włączani do badań klinicznych i nie obserwowano zmian w farmakokinetyce u tych pacjentów. Brak jest dostępnych danych dotyczących właściwości farmakokinetycznych u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby.
Dane niekliniczne wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, badań toksyczności po podaniu wielokrotnym oraz toksycznego wpływu na rozwój zarodka i płodu nie ujawniają szczególnego zagrożenia dla człowieka. Nie przeprowadzono badań rakotwórczego i mutagennego działania okrelizumabu.
W dwóch badaniach rozwoju przed- i pourodzeniowego u małp cynomolgus podawanie okrelizumabu od 20 dnia ciąży co najmniej do porodu wiązało się z występowaniem glomerulopatii, powstawaniem grudek chłonnych w szpiku kostnym, limfoplazmatycznym zapaleniem nerek oraz zmniejszoną masą jąder u potomstwa. Dawki podawane samicom w tych badaniach sprawiły, że maksymalne średnie stężenie w surowicy (Cmax) było 4,5- do 21-krotnie wyższe niż analogiczne stężenie przewidywane w warunkach klinicznych.
Zaobserwowano pięć przypadków stanu agonalnego noworodków: jeden przypisywany osłabieniu z powodu przedwczesnych narodzin i towarzyszącego mu oportunistycznego zakażenia bakteryjnego, jeden z powodu zakaźnego zapalenia opon mózgowych i mózgu z zajęciem móżdżku nowonarodzonego osobnika pochodzącego od matki z aktywnym zakażeniem (zapalenie sutka) i trzy z potwierdzeniem żółtaczki i uszkodzenia wątroby, z podejrzeniem etiologii wirusowej, prawdopodobnie poliomawirus. Na przebieg tych pięciu potwierdzonych lub podejrzewanych zakażeń mogła potencjalnie wpłynąć deplecja limfocytów B. U nowonarodzonego potomstwa samic narażonych na okrelizumab zauważono zmniejszoną populację limfocytów B w okresie pourodzeniowym.
Sodu octan trójwodny (E 262)
Kwas octowy lodowaty
Trehaloza dwuwodna
Polisorbat 20 (E 432)
Woda do wstrzykiwań
Nie obserwowano niezgodności pomiędzy tym produktem leczniczym a workami infuzyjnymi i zestawami do wlewu dożylnego wykonanymi z polichlorku winylu (PVC) lub poliolefiny (PO).
Tego produktu leczniczego nie wolno mieszać z innymi produktami leczniczymi, z wyjątkiem
produktów leczniczych wymienionych w punkcie 6.6.
Nieotwarta fiolka
2 lata
Rozcieńczony roztwór do wlewu dożylnego
Wykazano, że produkt zachowuje stabilność chemiczną i fizyczną przez 24 godziny w temperaturze 2- 8°C, a następnie przez 8 godzin w temperaturze pokojowej.
Z mikrobiologicznego punktu widzenia przygotowany roztwór do wlewu należy natychmiast zużyć. Jeśli roztwór nie zostanie natychmiast zużyty, za czas i warunki przechowywania produktu przed jego użyciem odpowiada użytkownik i zazwyczaj nie należy go przechowywać dłużej niż 24 godziny w temperaturze 2-8°C, a następnie 8 godzin w temperaturze pokojowej, chyba, że rozcieńczenia dokonano w kontrolowanych, zwalidowanych warunkach aseptycznych.
W przypadku, gdy wlew dożylny nie może być ukończony tego samego dnia, pozostałość roztworu
należy wyrzucić.
Przechowywać w lodówce (2°C – 8°C).
Nie zamrażać.
Fiolki należy przechowywać w zewnętrznym opakowaniu tekturowym w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rozcieńczeniu, patrz punkt 6.3.
10 ml koncentratu w fiolce (bezbarwne szkło typu I).
Opakowanie zawiera 1 lub 2 fiolki. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Instrukcja rozcieńczania
Produkt leczniczy powinien być przygotowywany przez osobę z fachowego personelu medycznego w sposób zapewniający aseptyczność. Nie wstrząsać fiolką. Do przygotowania rozcieńczonego roztworu do wlewu należy użyć jałowej igły i strzykawki.
Produkt jest przeznaczony wyłącznie do jednorazowego użycia.
Nie używać koncentratu, jeśli jest on przebarwiony lub jeśli koncentrat zawiera cząstki stałe (opis roztworu, patrz punkt 3).
Produkt leczniczy wymaga rozcieńczenia przed podaniem. Roztwory produktu leczniczego przeznaczone do podania dożylnego przygotowuje się przez rozcieńczenie koncentratu w worku infuzyjnym zawierającym izotoniczny roztwór chlorku sodu do infuzji o stężeniu 9 mg/ml (0,9%) (300 mg / 250 ml lub 600 mg / 500 ml), aby uzyskać docelowe stężenie okrelizumabu wynoszące około 1,2 mg/ml.
Rozcieńczony roztwór do wlewu należy podać za pomocą zestawu do infuzji wyposażonego w filtr przepływowy o średnicy porów 0,2 lub 0,22 m.
Przed rozpoczęciem wlewu dożylnego zawartość worka infuzyjnego powinna osiągnąć temperaturę pokojową.
Usuwanie
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Niemcy
EU/1/17/1231/001
EU/1/17/1231/002
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 8 stycznia 2018
Data ostatniego przedłużenia pozwolenia: 21 września 2022
Szczegółowe informacje o tym produkcie leczniczym są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu.
Powered by Biogen Poland Sp. z o.o.
(Biogen-101710)
 i
i  "Dodaj aplikację do ekranu początkowego"
"Dodaj aplikację do ekranu początkowego"